13.2
Two Families of Membrane Proteins Use ATP Hydrolysis to Pump Ions and Molecules Across Membranes
The extracellular fluid of animal cells has a salt concentration similar to that of seawater. However, cells must control their intracellular salt concentrations to facilitate specific processes, such as signal transduction and action potential propagation, and prevent unfavorable interactions with high concentrations of ions such as Ca2+. For instance, most animal cells contain a high concentration of K+ and a low concentration of Na+ relative to the external medium. These ionic gradients are generated by a specific transport system, an enzyme that is called the Na+–K+ pump or the Na+–K+ ATPase. The hydrolysis of ATP by the pump provides the energy needed for the active transport of Na+ out of the cell and K+ into the cell, generating the gradients. The pump is called the Na+–K+ ATPase because the hydrolysis of ATP takes place only when Na+ and K+ are present. This ATPase, like all such enzymes, requires Mg2+.
The change in free energy accompanying the transport of Na+ and K+ can be calculated. Suppose that the concentrations of Na+ outside and inside the cell are 143 and 14 mM, respectively, and the corresponding values for K+ are 4 and 157 mM. At a membrane potential of −50 mV and a temperature of 37°C, we can use the equation in the previous section to determine that the free-energy change in transporting 3 mol of Na+ out of the cell and 2 mol of K+ into the cell is 3(5.99) + 2(9.46) = +36.9 kJ mol−1 (+8.8 kcal mol−1). Under typical cellular conditions, the hydrolysis of a single ATP molecule per transport cycle provides sufficient free energy, about −50 kJ mol−1 (−12 kcal mol−1) to drive the uphill transport of these ions. The active transport of Na+ and K+ is of great physiological significance. Indeed, more than a third of the ATP consumed by a resting animal is used to pump these ions. The Na+–K+ gradient in animal cells controls cell volume, renders neurons and muscle cells electrically excitable, and drives the active transport of sugars and amino acids.
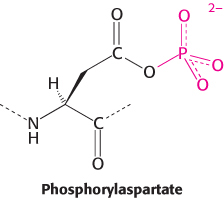
 The purification of other ion pumps has revealed a large family of evolutionarily related ion pumps including proteins from bacteria, archaea, and all eukaryotes. Each of these pumps is specific for a particular ion or set of ions. Two are of particular interest: the sarcoplasmic reticulum Ca2+ ATPase (or SERCA) transports Ca2+ out of the cytoplasm and into the sarcoplasmic reticulum of muscle cells, and the gastric H+–K+ ATPase is the enzyme responsible for pumping sufficient protons into the stomach to lower the pH to 1.0. These enzymes and the hundreds of known homologs, including the Na+–K+ ATPase, are referred to as P-type ATPases because they form a key phosphorylated intermediate. In the formation of this intermediate, a phosphoryl group from ATP is linked to the side chain of a specific conserved aspartate residue in the ATPase to form phosphorylaspartate.
The purification of other ion pumps has revealed a large family of evolutionarily related ion pumps including proteins from bacteria, archaea, and all eukaryotes. Each of these pumps is specific for a particular ion or set of ions. Two are of particular interest: the sarcoplasmic reticulum Ca2+ ATPase (or SERCA) transports Ca2+ out of the cytoplasm and into the sarcoplasmic reticulum of muscle cells, and the gastric H+–K+ ATPase is the enzyme responsible for pumping sufficient protons into the stomach to lower the pH to 1.0. These enzymes and the hundreds of known homologs, including the Na+–K+ ATPase, are referred to as P-type ATPases because they form a key phosphorylated intermediate. In the formation of this intermediate, a phosphoryl group from ATP is linked to the side chain of a specific conserved aspartate residue in the ATPase to form phosphorylaspartate.
P-type ATPases couple phosphorylation and conformational changes to pump calcium ions across membranes
Membrane pumps function by mechanisms that are simple in principle but often complex in detail. Fundamentally, each pump protein can exist in two principal conformational states, one with ion-binding sites open to one side of the membrane and the other with ion-binding sites open to the other side (Figure 13.2). To pump ions in a single direction across a membrane, the free energy of ATP hydrolysis must be coupled to the interconversion between these conformational states.
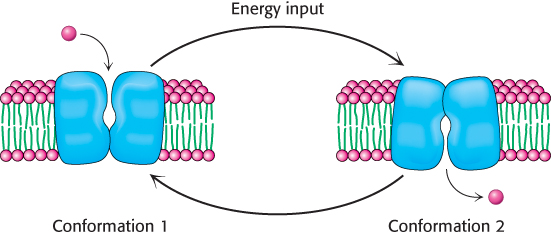
FIGURE 13.2Pump action. A simple scheme for the pumping of a molecule across a membrane. The pump interconverts to two conformational states, each with a binding site accessible to a different side of the membrane.
We will consider the structural and mechanistic features of P-type ATPases by examining SERCA. The properties of this P-type ATPase have been established in great detail by relying on crystal structures of the pump in five different states. This enzyme, which constitutes 80% of the protein in the sarcoplasmic reticulum membrane, plays an important role in relaxation of contracted muscle. Muscle contraction is triggered by an abrupt rise in the cytoplasmic calcium ion level. Subsequent muscle relaxation depends on the rapid removal of Ca2+ from the cytoplasm into the sarcoplasmic reticulum, a specialized compartment for Ca2+ storage, by SERCA. This pump maintains a Ca2+ concentration of approximately 0.1 μM in the cytoplasm compared with 1.5 mM in the sarcoplasmic reticulum.
The first structure of SERCA to be determined had Ca2+ bound, but no nucleotides present (Figure 13.3). SERCA is a single 110-kDa polypeptide with a transmembrane domain consisting of 10 α helices. The transmembrane domain includes sites for binding two calcium ions. Each calcium ion is coordinated to seven oxygen atoms coming from a combination of side-chain glutamate, aspartate, threonine, and asparagine residues, backbone carbonyl groups, and water molecules. A large cytoplasmic headpiece constitutes nearly half the molecular weight of the protein and consists of three distinct domains, each with a distinct function. One domain (N) binds the ATP nucleotide, another (P) accepts the phosphoryl group on a conserved aspartate residue, and the third (A) serves as an actuator, linking changes in the N and P domains to the transmembrane part of the enzyme.
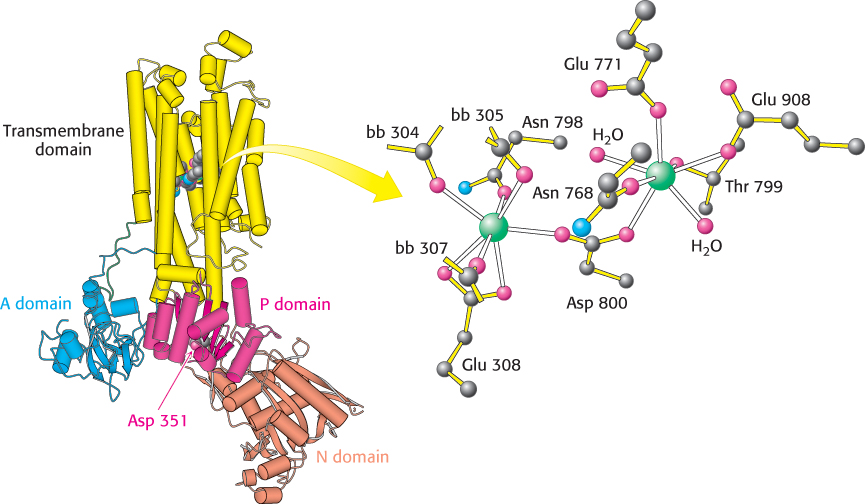
 FIGURE 13.3 Calcium-pump structure. The overall structure of the SERCA P-type ATPase. Notice the two calcium ions (green) that lie in the center of the transmembrane domain. A conserved aspartate residue (Asp 351) that binds a phosphoryl group lies in the P domain. The designation bb refers to backbone carbonyl groups. [Drawn from 1SU4.pdb.]
FIGURE 13.3 Calcium-pump structure. The overall structure of the SERCA P-type ATPase. Notice the two calcium ions (green) that lie in the center of the transmembrane domain. A conserved aspartate residue (Asp 351) that binds a phosphoryl group lies in the P domain. The designation bb refers to backbone carbonyl groups. [Drawn from 1SU4.pdb.]
SERCA is a remarkably dynamic protein. For example, the structure of SERCA without bound Ca2+, but with a phosphorylaspartate analog present in the P domain, is shown in Figure 13.4. The N and P domains are now closed around the phosphorylaspartate analog, and the A domain has rotated substantially relative to its position in SERCA with Ca2+ bound and without the phosphoryl analog. Furthermore, the transmembrane part of the enzyme has rearranged significantly and the well-organized Ca2+-binding sites are disrupted. These sites are now accessible from the side of the membrane opposite the N, P, and A domains.
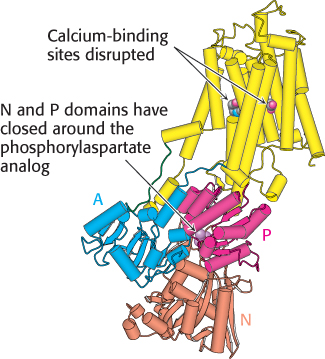
FIGURE 13.4 Conformational changes associated with calcium pumping. This structure was determined in the absence of bound calcium but with a phosphorylaspartate analog present in the P domain. Notice how different this structure is from the calcium-bound form shown in Figure 13.3: both the transmembrane part (yellow) and the A, P, and N domains have substantially rearranged. [Drawn from 1WPG.pdb.]
The structural results can be combined with other studies to construct a detailed mechanism for Ca2+ pumping by SERCA (Figure 13.5):
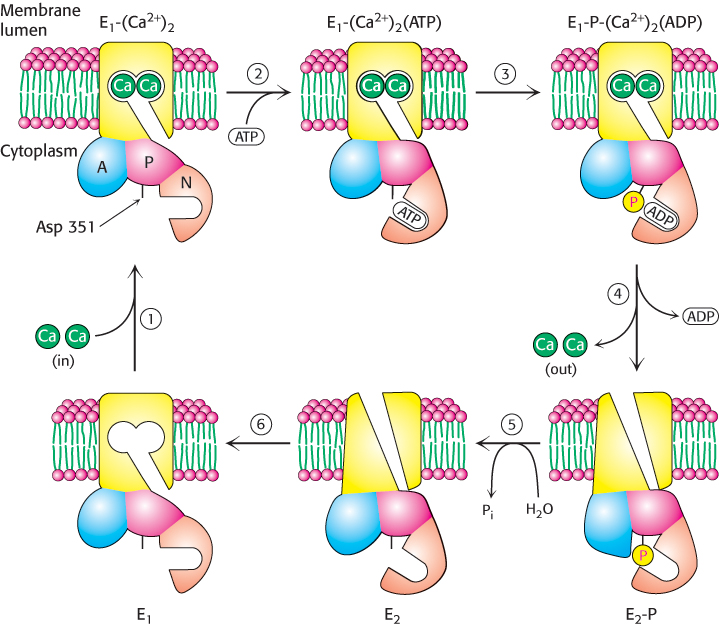
FIGURE 13.5Pumping calcium. Ca2+ATPase transports Ca2+ through the membrane by a mechanism that includes (1) Ca2+ binding from the cytoplasm, (2) ATP binding, (3) ATP cleavage with the transfer of a phosphoryl group to Asp 351 on the enzyme, (4) ADP release and eversion of the enzyme to release Ca2+ on the opposite side of the membrane, (5) hydrolysis of the phosphorylaspartate residue, and (6) eversion to prepare for the binding of Ca2+ from the cytoplasm.
1. The catalytic cycle begins with the enzyme in its unphosphorylated state with two calcium ions bound. We will refer to the overall enzyme confomation in this state as E1; with Ca2+ bound, it is E1-(Ca2+)2. In this conformation, SERCA can bind calcium ions only on the cytoplasmic side of the membrane. This conformation is shown in Figure 13.3.
2. In the E1 conformation, the enzyme can bind ATP. The N, P, and A domains undergo substantial rearrangement as they close around the bound ATP, but there is no substantial conformational change in the transmembrane domain. The calcium ions are now trapped inside the enzyme.
3. The phosphoryl group is then transferred from ATP to Asp 351.
4. Upon ADP release, the enzyme again changes its overall conformation, including the membrane domain this time. This new conformation is referred to as E2 or E2-P in its phosphorylated form. The process of interconverting the E1 and E2 conformations is sometimes referred to as eversion.
In the E2-P conformation, the Ca2+-binding sites become disrupted and the calcium ions are released to the side of the membrane opposite that at which they entered; ion transport has been achieved. This conformation is shown in Figure 13.4.
5. The phosphorylaspartate residue is hydrolyzed to release inorganic phosphate.
6. With the release of phosphate, the interactions stabilizing the E2 conformation are lost, and the enzyme everts to the E1 conformation.
The binding of two calcium ions from the cytoplasmic side of the membrane completes the cycle.
This mechanism likely applies to other P-type ATPases. For example, Na+–K+ ATPase is an α2β2 tetramer. Its α subunit is homologous to SERCA and includes a key aspartate residue analogous to Asp 351. The β subunit does not directly take part in ion transport. A mechanism analogous to that shown in Figure 13.5 applies, with three Na+ ions binding from the inside of the cell to the E1 conformation and two K+ ions binding from outside the cell to the E2 conformation.
Digitalis specifically inhibits the Na+–K+ pump by blocking its dephosphorylation
 Certain steroids derived from plants are potent inhibitors (Ki ≈ 10 nM) of the Na+–K+ pump. Digitoxigenin and ouabain are members of this class of inhibitors, which are known as cardiotonic steroids because of their strong effects on the heart (Figure 13.6). These compounds inhibit the dephosphorylation of the E2-P form of the ATPase when applied on the extracellular face of the membrane.
Certain steroids derived from plants are potent inhibitors (Ki ≈ 10 nM) of the Na+–K+ pump. Digitoxigenin and ouabain are members of this class of inhibitors, which are known as cardiotonic steroids because of their strong effects on the heart (Figure 13.6). These compounds inhibit the dephosphorylation of the E2-P form of the ATPase when applied on the extracellular face of the membrane.
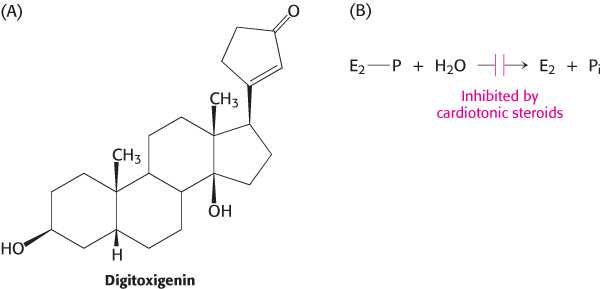
FIGURE 13.6Digitoxigenin. Cardiotonic steroids such as digitoxigenin inhibit the Na+–K+ pump by blocking the dephosphorylation of E2-P.
Digitalis is a mixture of cardiotonic steroids derived from the dried leaf of the foxglove plant (Digitalis purpurea). The compound increases the force of contraction of heart muscle and is consequently a choice drug in the treatment of congestive heart failure. Inhibition of the Na+–K+ pump by digitalis leads to a higher level of Na+ inside the cell. The diminished Na+ gradient results in slower extrusion of Ca2+ by the sodium–calcium exchanger, an antiporter (Section 13.3). The subsequent increase in the intracellular level of Ca2+ enhances the ability of cardiac muscle to contract. It is interesting to note that digitalis was used effectively long before the discovery of the Na+–K+ ATPase. In 1785, William Withering, a British physician, heard tales of an elderly woman, known as “the old woman of Shropshire,” who cured people of “dropsy” (which today would be recognized as congestive heart failure) with an extract of foxglove. Withering conducted the first scientific study of the effects of foxglove on congestive heart failure and documented its effectiveness.

Foxglove (Digitalis purpurea) is the source of digitalis, one of the most widely used drugs.
[Roger Hall/Shutterstock.]
Multidrug resistance highlights a family of membrane pumps with ATP-binding cassette domains
Studies of human disease revealed another large and important family of active-transport proteins, with structures and mechanisms quite different from those of the P-type ATPase family. These pumps were identified from studies on tumor cells in culture that developed resistance to drugs that had been initially quite toxic to the cells. Remarkably, the development of resistance to one drug had made the cells less sensitive to a range of other compounds. This phenomenon is known as multidrug resistance. In a significant discovery, the onset of multidrug resistance was found to correlate with the expression and activity of a membrane protein with an apparent molecular mass of 170 kDa. This protein acts as an ATP-dependent pump that extrudes a wide range of small molecules from cells that express it. The protein is called the multidrug-resistance (MDR) protein or P-glycoprotein (“glyco” because it includes a carbohydrate moiety). Thus, when cells are exposed to a drug, the MDR pumps the drug out of the cell before the drug can exert its effects.
Analysis of the amino acid sequences of MDR and homologous proteins revealed a common architecture (Figure 13.7A). Each protein comprises four domains: two membrane-spanning domains and two ATP-binding domains. The ATP-binding domains of these proteins are called ATP-binding cassettes (ABCs) and are homologous to domains in a large family of transport proteins in bacteria and archaea. Transporters that include these domains are called ABC transporters. With 79 members, the ABC transporters are the largest single family identified in the E. coli genome. The human genome includes more than 150 ABC transporter genes.
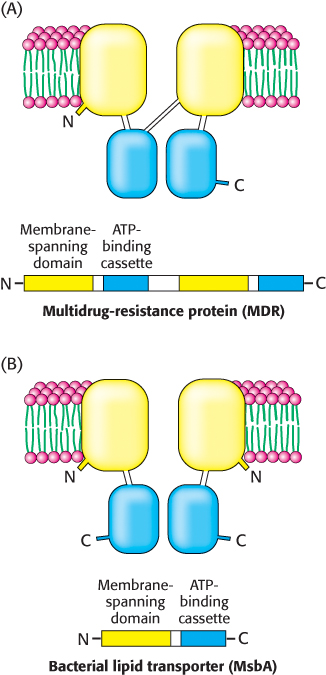
FIGURE 13.7Domain arrangement of ABC transporters. ABC transporters are a large family of homologous proteins composed of two transmembrane domains and two ATP-binding domains called ATP-binding cassettes (ABCs). (A) The multidrug-resistance protein is a single polypeptide chain containing all four domains, whereas (B) the bacterial lipid transporter MsbA consists of a dimer of two identical chains, containing one of each domain.
The ABC proteins are members of the P-loop NTPase superfamily (Section 9.4). The three-dimensional structures of several members of the ABC transporter family have now been determined, including that of the bacterial lipid transporter MsbA. In contrast with the eukaryotic MDR protein, this protein is a dimer of 62-kDa chains: the amino-terminal half of each protein contains the membrane-spanning domain, and the carboxyl-terminal half contains the ATP-binding cassette (Figure 13.7B). Prokaryotic ABC proteins are often made up of multiple subunits, such as a dimer of identical chains, as above, or as a heterotetramer of two membrane-spanning domain subunits and two ATP-binding-cassette subunits. The consolidation of the enzymatic activities of several polypeptide chains in prokaryotes to a single chain in eukaryotes is a theme that we will see again. The two ATP-binding cassettes are in contact, but they do not interact strongly in the absence of bound ATP (Figure 13.8). On the basis of this structure and others, as well as on other experiments, a mechanism for active transport by these proteins has been developed (Figure 13.9):
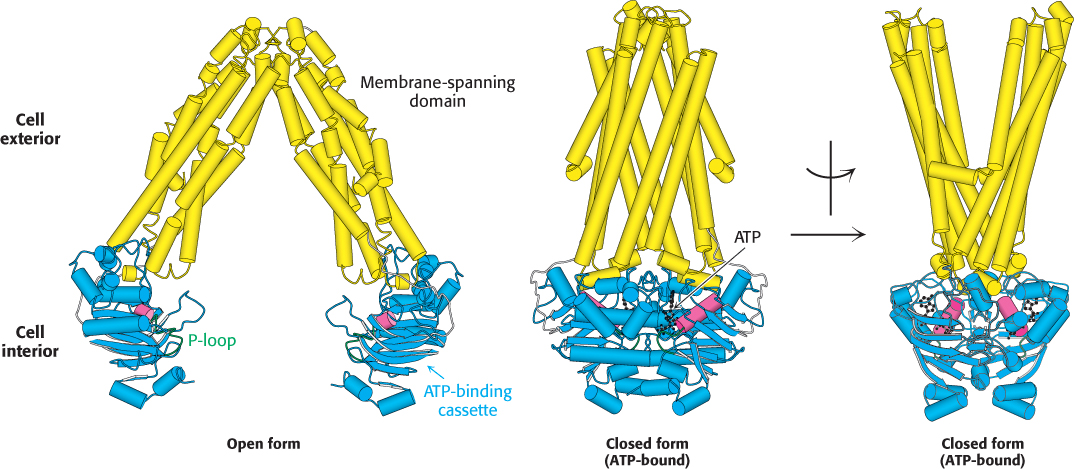
FIGURE 13.8ABC transporter structure. Two structures of the bacterial lipid transporter MsbA, a representative ABC transporter. The nucleotide-free, inward-facing form is on the left and the ATP-bound, outward-facing form is shown in two views (rotated by 90 degrees) in the center and on the right. The two ATP-binding cassettes (blue) are related to the P-loop NTPases and, like them, contain P-loops (green). The α helix adjacent to the P-loop is shown in red. [Drawn from 3B5W and 3B60.pdb.]
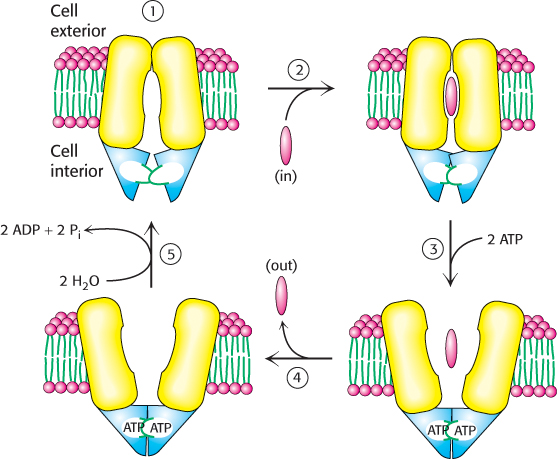
FIGURE 13.9ABC transporter mechanism. The mechanism includes the following steps: (1) opening of the channel toward the inside of the cell, (2) substrate binding and conformational changes in the ATP-binding cassettes, (3) ATP binding and opening of the channel to the opposite face of the membrane, (4) release of the substrate to the outside of the cell, and (5) ATP hydrolysis to reset the transporter to its initial state.
1. The catalytic cycle begins with the transporter free of both ATP and substrate. While the distance between the ATP-binding cassettes in this form may vary with the individual transporter, the substrate binding region of the transporter faces inward.
2. Substrate enters the central cavity of the transporter from inside the cell. Substrate binding induces conformational changes in the ATP-binding cassettes that increase their affinity for ATP.
3. ATP binds to the ATP-binding cassettes, changing their conformations so that the two domains interact strongly with one another. The close interaction of the ABCs reorients the transmembrane helices such that the substrate binding site is now facing outside the cell (Figure 13.8, far right).
4. The outward facing conformation of the transporter has reduced affinity for the substrate, enabling the release of the substrate on the opposite face of the membrane.
5. The hydrolysis of ATP and the release of ADP and inorganic phosphate reset the transporter for another cycle.
Whereas eukaryotic ABC transporters generally act to export molecules from inside the cell, prokaryotic ABC transporters often act to import specific molecules from outside the cell. A specific binding protein acts in concert with the bacterial ABC transporter, delivering the substrate to the transporter and stimulating ATP hydrolysis inside the cell. These binding proteins are present in the periplasm, the compartment between the two membranes that surround some bacterial cells (Figure 12.34A).
Thus, ABC transporters use a substantially different mechanism from the P-type ATPases to couple the ATP hydrolysis reaction to conformational changes. Nonetheless, the net result is the same: the transporters are converted from one conformation capable of binding substrate from one side of the membrane to another that releases the substrate on the other side.