Ubiquitin tags proteins for destruction
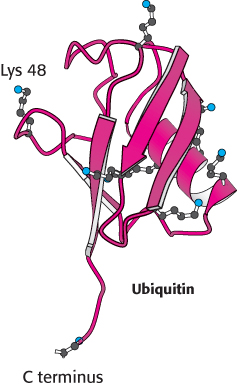
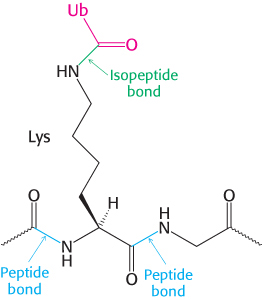
Ubiquitin is highly conserved in eukaryotes: yeast and human ubiquitin differ at only 3 of 76 residues. The carboxyl-terminal glycine residue of ubiquitin becomes covalently attached to the ε-amino groups of several lysine residues on a protein destined to be degraded. The energy for the formation of these isopeptide bonds (iso because ε- rather than α-amino groups are targeted) comes from ATP hydrolysis.
Three enzymes participate in the attachment of ubiquitin to a protein (Figure 23.3): ubiquitin-activating enzyme, or E1; ubiquitin-conjugating enzyme, or E2; and ubiquitin–protein ligase, or E3. First, the C-terminal carboxylate group of ubiquitin becomes linked to a sulfhydryl group of E1 by a thioester bond. This ATP-driven reaction is reminiscent of fatty acid activation (Section 22.2). In this reaction, an acyl adenylate is formed at the C-terminal carboxylate of ubiquitin with the release of pyrophosphate, and the ubiquitin is subsequently transferred to a sulfhydryl group of a key cysteine residue in E1. The activated ubiquitin is then shuttled to a sulfhydryl group of E2, a reaction catalyzed by E2 itself. Finally, E3 catalyzes the transfer of ubiquitin from E2 to an ε-amino group on the target protein. The ubiquitination reaction is processive: E3 remains bound to the target proteins and generates a chain of ubiquitin molecules by linking the ε-amino group of lysine residue 48 of one ubiquitin molecule to the terminal carboxylate of another. A chain of four or more ubiquitin molecules is especially effective in signaling the need for degradation (Figure 23.4).
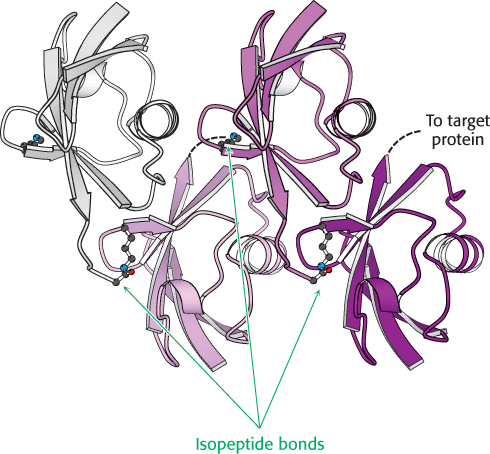
FIGURE 23.4Structure of tetraubiquitin. Four ubiquitin molecules are linked by isopeptide bonds. Notice that each isopeptide bond is formed by the linkage of the carboxylate group at the end of the extended C terminus with the ε-amino group of a lysine residue. Dashed lines indicate the positions of the extended C-termini that were not observed in the crystal structure. This unit is the primary signal for degradation when linked to a target protein.
[Drawn from 1TBE. pdb.]
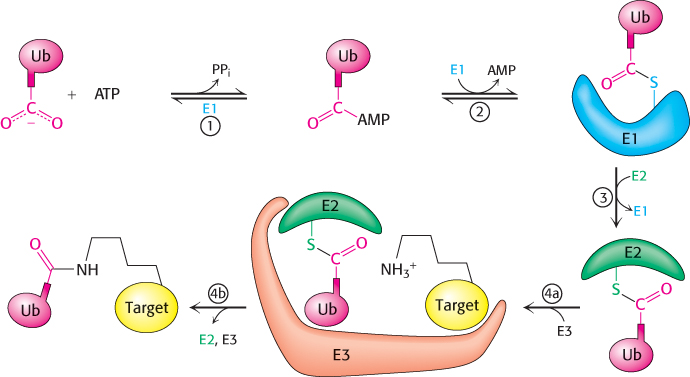
FIGURE 23.3Ubiquitin conjugation. The ubiquitin-activating enzyme E1 adenylates ubiquitin (Ub) (1) and transfers the ubiquitin to one of its own cysteine residues (2). Ubiquitin is then transferred to a cysteine residue in the ubiquitin-conjugating enzyme E2 by the E2 enzyme. (3). Finally, the ubiquitin–protein ligase E3 transfers the ubiquitin to a lysine residue on the target protein (4a and 4b).
TABLE 23.2 Dependence of the half-lives of cytoplasmic yeast proteins on the identity of their amino-terminal residues
Highly stabilizing residues (t1/2 > 20 hours) |
|
Pro
|
Ser
|
Thr
|
Val
|
Intrinsically destabilizing residues (t1/2 = 2 to 30 minutes) |
|
Lys
|
Phe
|
Trp
|
Tyr
|
Destabilizing residues after chemical modification (t1/2 = 3 to 30 minutes) |
|
|
|
|
|
Data from J. W. Tobias, T. E. Schrader, G. Rocap, and A. Varshavsky. Science 254(1991):1374–1377. |
What determines whether a protein becomes ubiquitinated? A specific sequence of amino acids, termed a degron, indicates that a protein should be degraded. One such signal appears to be quite simple. The half-life of a cytoplasmic protein is determined to a large extent by its amino-terminal residue (Table 23.2). This dependency is referred to as the N-terminal rule or the N-terminal degron. A yeast protein with methionine at its N terminus typically has a half-life of more than 20 hours, whereas one with arginine at this position has a half-life of about 2 minutes. A highly destabilizing N-terminal residue such as arginine or leucine favors rapid ubiquitination, whereas a stabilizing residue such as methionine or proline does not. Why is the N-terminal rule only apparently simple? Sometimes the N-terminal degron is exposed only after the protein is proteolytically cleaved. Such degrons are called pro-N-terminal degrons, in analogy to proenzymes (Section 10.4), because the protein must be cleaved to expose the signal. In other cases, the destabilizing amino acid is added to the protein after the protein is synthesized. Other modifications, notably N-terminal acetylation, can also activate a degron. Additional degrons thought to identify proteins for degradation include cyclin destruction boxes, which are amino acid sequences that mark cell-cycle proteins for destruction, and PEST sequences, which contain the amino acid sequence proline (P, single-letter abbreviation), glutamic acid (E), serine (S), and threonine (T).
E3 enzymes are the readers of N-terminal residues. Although most eukaryotes have only one or a small number of distinct E1 enzymes, all eukaryotes have many distinct E2 and E3 enzymes. Moreover, there appears to be only a single family of evolutionarily related E2 proteins but three distinct families of E3 proteins, all together consisting of hundreds of members. Indeed, the E3 family is one of the largest gene families in human beings. The diversity of target proteins that must be tagged for destruction requires a large number of E3 proteins as readers.
 Three examples demonstrate the importance of E3 proteins to normal cell function. Proteins that are not broken down owing to a defective E3 may accumulate to create a disease of protein aggregation such as juvenile or early-onset Parkinson disease. A defect in another member of the E3 family causes Angelman syndrome, a severe neurological disorder characterized by mental retardation, absence of speech, uncoordinated movement, and hyperactivity. Conversely, uncontrolled protein turnover also can create dangerous pathological conditions. For example, human papillomavirus (HPV) encodes a protein that activates a specific E3 enzyme. The enzyme ubiquitinates the tumor suppressor p53 and other proteins that control DNA repair, which are then destroyed. The activation of this E3 enzyme is observed in more than 90% of cervical carcinomas. Thus, the inappropriate marking of key regulatory proteins for destruction can trigger further events, leading to tumor formation.
Three examples demonstrate the importance of E3 proteins to normal cell function. Proteins that are not broken down owing to a defective E3 may accumulate to create a disease of protein aggregation such as juvenile or early-onset Parkinson disease. A defect in another member of the E3 family causes Angelman syndrome, a severe neurological disorder characterized by mental retardation, absence of speech, uncoordinated movement, and hyperactivity. Conversely, uncontrolled protein turnover also can create dangerous pathological conditions. For example, human papillomavirus (HPV) encodes a protein that activates a specific E3 enzyme. The enzyme ubiquitinates the tumor suppressor p53 and other proteins that control DNA repair, which are then destroyed. The activation of this E3 enzyme is observed in more than 90% of cervical carcinomas. Thus, the inappropriate marking of key regulatory proteins for destruction can trigger further events, leading to tumor formation.
It is important to note that the role of ubiquitin is much broader than merely marking proteins for destruction. Although we have focused on protein degradation, monoubiquitination also regulates proteins involved in DNA repair, chromatin remodeling, and protein kinase activation, among other biochemical processes.
The proteasome digests the ubiquitin-tagged proteins
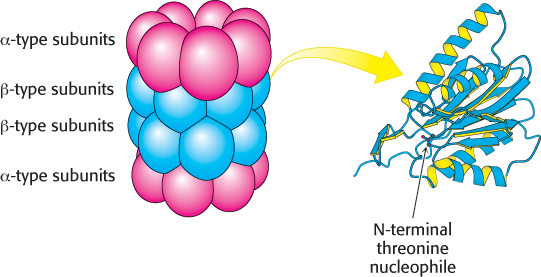
FIGURE 23.520S proteasome. The 20S proteasome comprises 28 homologous subunits (α-type, red; β-type, blue), arranged in four rings of 7 subunits each. Some of the β-type subunits (right) include protease active sites at their amino termini.
[Subunit drawn from 1RYP.pdb.]
If ubiquitin is the mark of death, what is the executioner? A large protease complex called the proteasome or the 26S proteasome digests the ubiquitinated proteins. This ATP-driven multisubunit protease spares ubiquitin, which is then recycled. The 26S proteasome is a complex of two components: a 20S catalytic unit and a 19S regulatory unit.
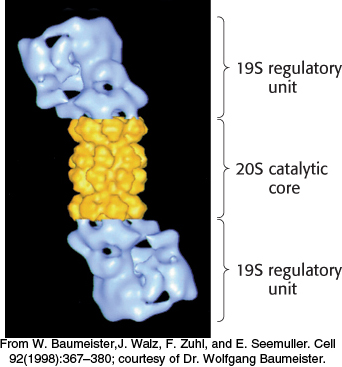
FIGURE 23.626S proteasome. A 19S regulatory subunit is attached to each end of the 20S catalytic unit.
[From W. Baumeister, J. Walz, F. Zuhl, and E. Seemuller. Cell 92(1998):367–380; courtesy of Dr. Wolfgang Baumeister.]
The 20S unit is composed of 28 subunits, encoded by 14 genes, arranged into 4 heteroheptomeric rings stacked to form a structure resembling a barrel (Figure 23.5). The outer two rings of the barrel are made up of α-type subunits and the inner two rings of β-type subunits. The 20S catalytic core is a sealed barrel. Access to its interior is controlled by a 19S regulatory unit, itself a 700-kDa complex made up of 19 subunits. Two such 19S complexes bind to the 20S proteasome core, one at each end, to form the complete 26S proteasome (Figure 23.6). The 19S regulatory unit has three functions. First, components of the 19S unit are ubiquitin receptors that bind specifically to polyubiquitin chains, thereby ensuring that only ubiquitinated proteins are degraded. Second, an isopeptidase in the 19S unit cleaves off intact ubiquitin molecules from the proteins so that they can be reused. Finally, the doomed protein is unfolded and directed into the catalytic core. Key components of the 19S complex are six ATPases of a type called the AAA class (ATPase associated with various cellular activities). ATP hydrolysis assists the 19S complex to unfold the substrate and induce conformational changes in the 20S catalytic core so that the substrate can be passed into the center of the complex.
The proteolytic active sites are sequestered in the interior of the barrel to protect potential substrates until they are directed into the barrel. There are three types of active sites in the β subunits, each with a different specificity, but all employ an N-terminal threonine. The hydroxyl group of the threonine residue is converted into a nucleophile that attacks the carbonyl groups of peptide bonds to form acyl-enzyme intermediates. Substrates are degraded in a processive manner without the release of degradation intermediates, until the substrate is reduced to peptides ranging in length from seven to nine residues. These peptide products are released from the proteasome and further degraded by other cellular proteases to yield individual amino acids. Thus, the ubiquitination pathway and the proteasome cooperate to degrade unwanted proteins. Figure 23.7 presents an overview of the fates of amino acids following proteasomal digestion.
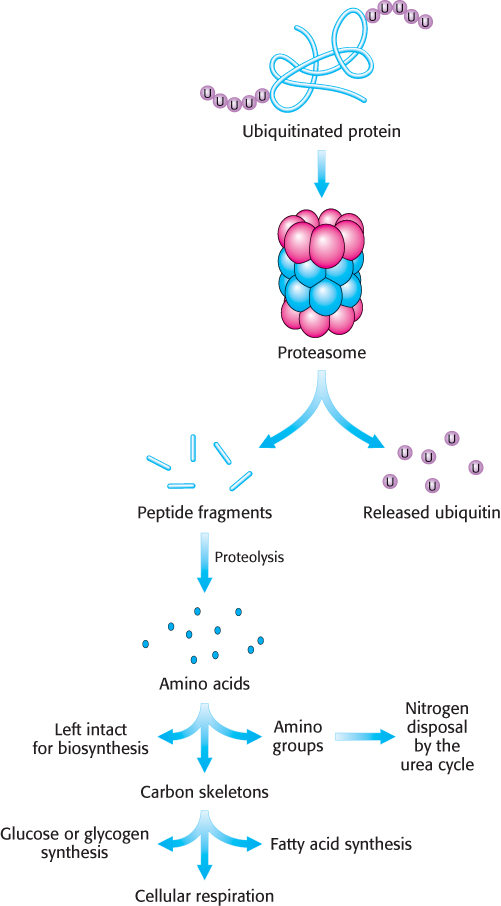
FIGURE 23.7The proteasome and other proteases generate free amino acids. Ubiquitinated proteins are processed to peptide fragments from which the ubiquitin is subsequently removed and recycled. The peptide fragments are further digested to yield free amino acids, which can be used for biosynthetic reactions, most notably protein synthesis. Alternatively, the amino group can be removed and processed to urea and the carbon skeleton can be used to synthesize carbohydrate or fats or used directly as a fuel for cellular respiration.
The ubiquitin pathway and the proteasome have prokaryotic counterparts
 Both the ubiquitin pathway and the proteasome appear to be present in all eukaryotes. Homologs of the proteasome are also found in some prokaryotes. The proteasomes of some archaea are quite similar in overall structure to their eukaryotic counterparts and similarly have 28 subunits (Figure 23.8). In the archaeal proteasome, however, all α outer-ring subunits and all β inner-ring subunits are identical; in eukaryotes, each α or β subunit is one of seven different isoforms. This specialization provides distinct substrate specificity.
Both the ubiquitin pathway and the proteasome appear to be present in all eukaryotes. Homologs of the proteasome are also found in some prokaryotes. The proteasomes of some archaea are quite similar in overall structure to their eukaryotic counterparts and similarly have 28 subunits (Figure 23.8). In the archaeal proteasome, however, all α outer-ring subunits and all β inner-ring subunits are identical; in eukaryotes, each α or β subunit is one of seven different isoforms. This specialization provides distinct substrate specificity.
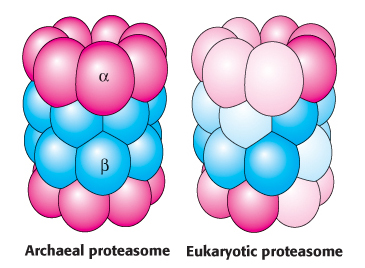
FIGURE 23.8Proteasome evolution. The archaeal proteasome consists of 14 identical α subunits and 14 identical β subunits. In the eukaryotic proteasome, gene duplication and specialization has led to 7 distinct subunits of each type. The overall architecture of the proteasome is conserved.
Protein degradation can be used to regulate biological function
TABLE 23.3 Processes regulated by protein degradation
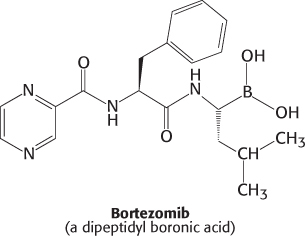
Table 23.3 lists a number of physiological processes that are controlled at least in part by protein degradation through the ubiquitin–proteasome pathway. In each case, the proteins being degraded are regulatory proteins. Consider, for example, control of the inflammatory response. A transcription factor called NF-κB (NF for nuclear factor) initiates the expression of a number of the genes that take part in this response. This factor is itself activated by the degradation of an attached inhibitory protein, I-κB (I for inhibitor). In response to inflammatory signals that bind to membrane-bound receptors, I-κB is phosphorylated at two serine residues, creating an E3 binding site. The binding of E3 leads to the ubiquitination and degradation of I-κB, unleashing NF-κB. The liberated transcription factor migrates from the cytoplasm to the nucleus to stimulate the transcription of the target genes. The NF-κB–I-κB system illustrates the interplay of several key regulatory motifs: receptor-mediated signal transduction, phosphorylation, compartmentalization, controlled and specific degradation, and selective gene expression. The importance of the ubiquitin–proteasome system for the regulation of gene expression is highlighted by the use of bortezomib (Velcade), a potent inhibitor of the proteasome, as a therapy for multiple myeloma. Bortezomib is a dipeptidyl boronic acid inhibitor of the proteasome.
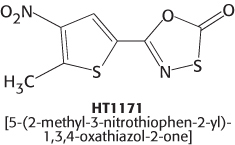
The evolutionary studies of proteasomes described previously have also yielded potential clinical benefits. The bacterial pathogen Mycobacterium tuberculosis, the cause of tuberculosis, harbors a proteasome that is very similar to the human counterpart. Nevertheless, recent work has shown that it is possible to exploit the differences between the human and the bacterial proteasomes to develop specific inhibitors of the M. tuberculosis complex. Oxathiazol-2-one compounds such as HT1171 are suicide inhibitors (Section 8.5) of the proteolytic activity of the M. tuberculosis proteasome, but have no effect on the proteasomes of the human host. This is especially exciting because these drugs kill the nonreplicating form of M. tuberculosis, and thus may not require the prolonged treatment required with conventional drugs, thereby reducing the likelihood of drug resistance due to interruption of the treatment regime.