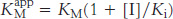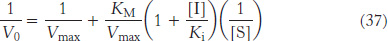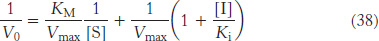8.5
Enzymes Can Be Inhibited by Specific Molecules
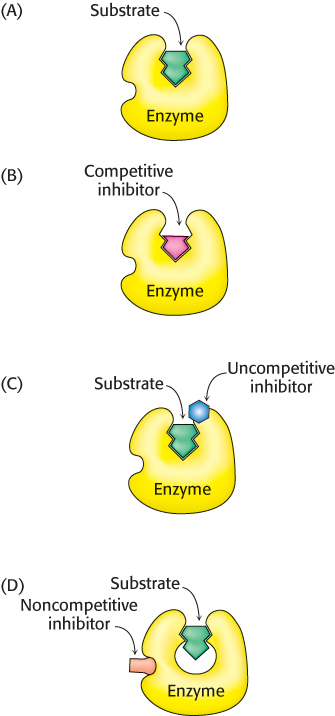
FIGURE 8.14Distinction between reversible inhibitors. (A) Enzyme–substrate complex; (B) a competitive inhibitor binds at the active site and thus prevents the substrate from binding; (C) an uncompetitive inhibitor binds only to the enzyme–substrate complex; (D) a noncompetitive inhibitor does not prevent the substrate from binding.
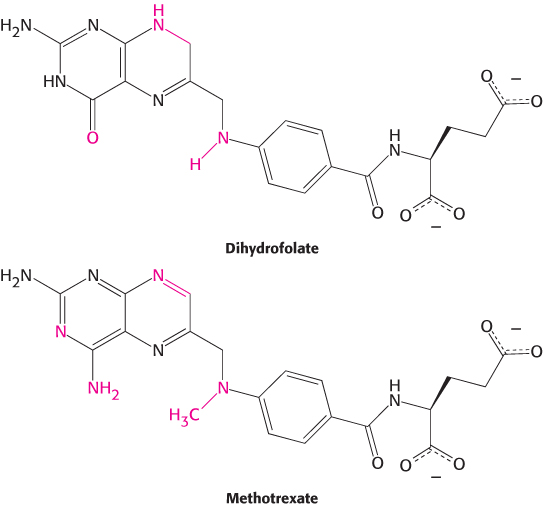
FIGURE 8.15Enzyme inhibitors. The substrate dihydrofolate and its structural analog methotrexate. Regions with structural differences are shown in red.
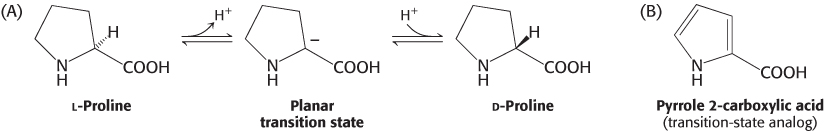
The activity of many enzymes can be inhibited by the binding of specific small molecules and ions. This means of inhibiting enzyme activity serves as a major control mechanism in biological systems, typified by the regulation of allosteric enzymes. In addition, many drugs and toxic agents act by inhibiting enzymes (Chapter 36). This type of enzyme inhibition is not usually the result of evolutionary forces, as it is for allosteric enzymes, but rather due to design of inhibitors by scientists or simple chance discovery of inhibitory molecules. Examining inhibition can be a source of insight into the mechanism of enzyme action: specific inhibitors can often be used to identify residues critical for catalysis. Transition-state analogs are especially potent inhibitors.
Enzyme inhibition can be either irreversible or reversible. An irreversible inhibitor dissociates very slowly from its target enzyme because it has become tightly bound to the enzyme, either covalently or noncovalently. Some irreversible inhibitors are important drugs. Penicillin acts by covalently modifying the enzyme transpeptidase, thereby preventing the synthesis of bacterial cell walls and thus killing the bacteria. Aspirin acts by covalently modifying the enzyme cyclooxygenase, reducing the synthesis of signaling molecules in inflammation.
Reversible inhibition, in contrast with irreversible inhibition, is characterized by a rapid dissociation of the enzyme–inhibitor complex. In the type of reversible inhibition called competitive inhibition, an enzyme can bind substrate (forming an ES complex) or inhibitor (EI) but not both (ESI, enzyme-substrate-inhibitor complex). The competitive inhibitor often resembles the substrate and binds to the active site of the enzyme (Figure 8.14). The substrate is thereby prevented from binding to the same active site. A competitive inhibitor diminishes the rate of catalysis by reducing the proportion of enzyme molecules bound to a substrate. At any given inhibitor concentration, competitive inhibition can be relieved by increasing the substrate concentration. Under these conditions, the substrate successfully competes with the inhibitor for the active site. Methotrexate is an especially potent competitive inhibitor of the enzyme dihydrofolate reductase, which plays a role in the biosynthesis of purines and pyrimidines. Methotrexate is a structural analog of dihydrofolate, a substrate for dihydrofolate reductase (Figure 8.15). What makes it such a potent competitive inhibitor is that it binds to the enzyme 1000 times as tightly as the natural substrate binds, and it inhibits nucleotide base synthesis. It is used to treat cancer (Section 25.3).
Uncompetitive inhibition is essentially substrate-dependent inhibition in that the inhibitor binds only to the enzyme–substrate complex. The binding site of an uncompetitive inhibitor is created only on interaction of the enzyme and substrate (Figure 8.14C). Uncompetitive inhibition cannot be overcome by the addition of more substrate.
In noncompetitive inhibition, the inhibitor and substrate can bind simultaneously to an enzyme molecule at different binding sites (Figure 8.14D). Unlike uncompetitive inhibition, a noncompetitive inhibitor can bind free enzyme or the enzyme–substrate complex. A noncompetitive inhibitor acts by decreasing the concentration of functional enzyme rather than by diminishing the proportion of enzyme molecules that are bound to substrate. The net effect is to decrease the turnover number. Noncompetitive inhibition, like uncompetitive inhibition, cannot be overcome by increasing the substrate concentration. A more complex pattern, called mixed inhibition, is produced when a single inhibitor both hinders the binding of substrate and decreases the turnover number of the enzyme.
The different types of reversible inhibitors are kinetically distinguishable
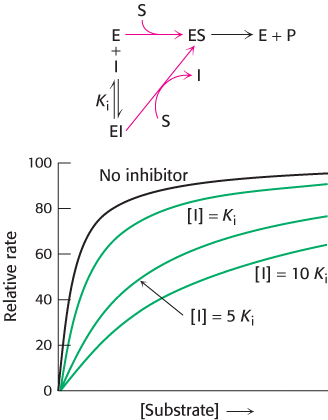
FIGURE 8.16Kinetics of a competitive inhibitor. As the concentration of a competitive inhibitor increases, higher concentrations of substrate are required to attain a particular reaction velocity. The reaction pathway suggests how sufficiently high concentrations of substrate can completely relieve competitive inhibition.
How can we determine whether a reversible inhibitor acts by competitive, uncompetitive, or noncompetitive inhibition? Let us consider only enzymes that exhibit Michaelis–Menten kinetics. Measurements of the rates of catalysis at different concentrations of substrate and inhibitor serve to distinguish the three types of inhibition. In competitive inhibition, the inhibitor competes with the substrate for the active site. The dissociation constant for the inhibitor is given by
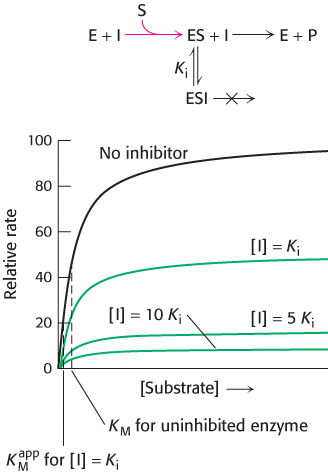
FIGURE 8.17Kinetics of an uncompetitive inhibitor. The reaction pathway shows that the inhibitor binds only to the enzyme–substrate complex. Consequently, Vmax cannot be attained, even at high substrate concentrations. The apparent value for KM is lowered, becoming smaller as more inhibitor is added.
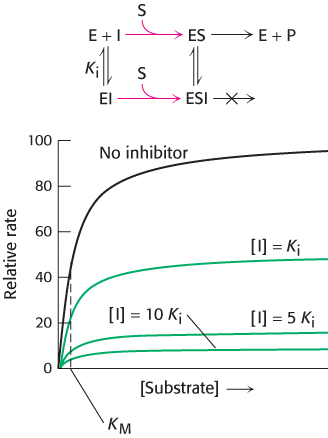
FIGURE 8.18Kinetics of a noncompetitive inhibitor. The reaction pathway shows that the inhibitor binds both to free enzyme and to an enzyme–substrate complex. Consequently, as with uncompetitive competition, Vmax cannot be attained. In pure noncompetitive inhibition, KM remains unchanged, and so the reaction rate increases more slowly at low substrate concentrations than is the case for uncompetitive competition.
The smaller the Ki, the more potent the inhibition. The hallmark of competitive inhibition is that it can be overcome by a sufficiently high concentration substrate (Figure 8.16). The effect of a competitive inhibitor is to increase the apparent value of KM, meaning that more substrate is needed to obtain the same reaction rate. This new value of KM, called 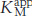 is numerically equal to
is numerically equal to
where [I] is the concentration of inhibitor and Ki is the dissociation constant for the enzyme–inhibitor complex. In the presence of a competitive inhibitor, an enzyme will have the same Vmax as in the absence of an inhibitor. At a sufficiently high concentration, virtually all the active sites are filled with substrate, and the enzyme is fully operative.
Competitive inhibitors are commonly used as drugs. Drugs such as ibuprofen are competitive inhibitors of enzymes that participate in signaling pathways in the inflammatory response. Statins are drugs that reduce high cholesterol levels by competitively inhibiting a key enzyme in cholesterol biosynthesis (Section 26.3).
In uncompetitive inhibition, the inhibitor binds only to the ES complex. This enzyme–substrate–inhibitor complex, ESI, does not go on to form any product. Because some unproductive ESI complex will always be present, Vmax will be lower in the presence of inhibitor than in its absence (Figure 8.17). The uncompetitive inhibitor lowers the apparent value of KM because the inhibitor binds to ES to form ESI, depleting ES. To maintain the equilibrium between E and ES, more S binds to E, increasing the apparent value of k1 and thereby reducing the apparent value of KM (see equation 18). Thus, a lower concentration of S is required to form half of the maximal concentration of ES. The herbicide glyphosate, also known as Roundup, is an uncompetitive inhibitor of an enzyme in the biosynthetic pathway for aromatic amino acids.
In noncompetitive inhibition (Figure 8.18), substrate can bind either to the enzyme or the enzyme–inhibitor complex. However, the enzyme–inhibitor–substrate complex does not proceed to form product. In pure noncompetitive inhibition, the Ki for the inhibitor binding to E is the same as for binding to ES complex. The value of Vmax is decreased to a new value called 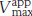 , whereas the value of KM is unchanged. The maximal velocity in the presence of a pure noncompetitive inhibitor,
, is given by
, whereas the value of KM is unchanged. The maximal velocity in the presence of a pure noncompetitive inhibitor,
, is given by

Why is Vmax lowered though KM remains unchanged? In essence, the inhibitor simply lowers the concentration of functional enzyme. The resulting solution behaves as a more dilute solution of enzyme does. Noncompetitive inhibition cannot be overcome by increasing the substrate concentration. Doxycycline, an antibiotic, functions at low concentrations as a noncompetitive inhibitor of a proteolytic enzyme (collagenase). It is used to treat periodontal disease. Some of the toxic effects of lead poisoning may be due to lead’s ability to act as a noncompetitive inhibitor of a host of enzymes. Lead reacts with crucial sulfhydryl groups in these enzymes.
Double-reciprocal plots are especially useful for distinguishing between competitive, uncompetitive, and noncompetitive inhibitors. In competitive inhibition, the intercept on the y-axis of the plot of 1/V0 versus 1/[S] is the same in the presence and in the absence of inhibitor, although the slope is increased (Figure 8.19). The intercept is unchanged because a competitive inhibitor does not alter Vmax. The increase in the slope of the 1/V0 versus 1/[S] plot indicates the strength of binding of a competitive inhibitor. In the presence of a competitive inhibitor, equation 27 is replaced by
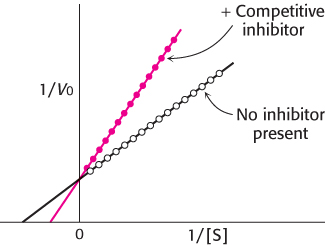
FIGURE 8.19Competitive inhibition illustrated on a double-reciprocal plot. A double-reciprocal plot of enzyme kinetics in the presence and absence of a competitive inhibitor illustrates that the inhibitor has no effect on Vmax but increases KM.
In other words, the slope of the plot is increased by the factor (1 + [I]/Ki) in the presence of a competitive inhibitor. Consider an enzyme with a KM of 10−4 M. In the absence of inhibitor, when V0 = Vmax/2 when [S] = 10−4 M. In the presence of a 2 × 10−3 M competitive inhibitor that is bound to the enzyme with a Ki of 10−3 M, the apparent KM 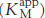 will be equal to KM(1 + [I]/Ki), or 3 × 10−4 M. Substitution of these values into equation 37 gives when V0 = Vmax/4, when [S] = 10−4 M. The presence of the competitive inhibitor thus cuts the reaction rate in half at this substrate concentration.
will be equal to KM(1 + [I]/Ki), or 3 × 10−4 M. Substitution of these values into equation 37 gives when V0 = Vmax/4, when [S] = 10−4 M. The presence of the competitive inhibitor thus cuts the reaction rate in half at this substrate concentration.
In uncompetitive inhibition (Figure 8.20), the inhibitor combines only with the enzyme–substrate complex. The equation that describes the double-reciprocal plot for an uncompetitive inhibitor is
The slope of the line, KM/Vmax, is the same as that for the uninhibited enzyme, but the intercept on the y-axis will be increased by 1 + [I]/Ki. Consequently, the lines in double-reciprocal plots will be parallel.
In pure noncompetitive inhibition (Figure 8.21), the inhibitor can combine with either the enzyme or the enzyme–substrate complex with the same dissociation constant. The value of Vmax is decreased to the new value
, and so the intercept on the vertical axis is increased (equation 36). The new slope, which is equal to KM/
, is larger by the same factor. In contrast with Vmax, KM is not affected by pure noncompetitive inhibition.
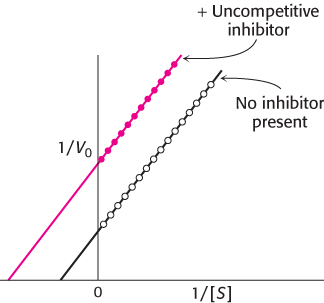
FIGURE 8.20Uncompetitive inhibition illustrated by a double-reciprocal plot. An uncompetitive inhibitor does not affect the slope of the double-reciprocal plot. Vmax and KM are reduced by equivalent amounts.
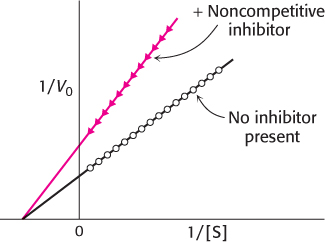
FIGURE 8.21Noncompetitive inhibition illustrated on a double-reciprocal plot. A double-reciprocal plot of enzyme kinetics in the presence and absence of a pure noncompetitive inhibitor shows that KM is unaltered and Vmax is decreased.
Irreversible inhibitors can be used to map the active site
In Chapter 9, we will examine the chemical details of how enzymes function. The first step in obtaining the chemical mechanism of an enzyme is to determine what functional groups are required for enzyme activity. How can we ascertain what these functional groups are? X-ray crystallography of the enzyme bound to its substrate or substrate analog provides one approach. Irreversible inhibitors that covalently bond to the enzyme provide an alternative and often complementary approach: the inhibitors modify the functional groups, which can then be identified. Irreversible inhibitors can be divided into three categories: group-specific reagents, reactive substrate analogs (also called affinity labels), and suicide inhibitors.
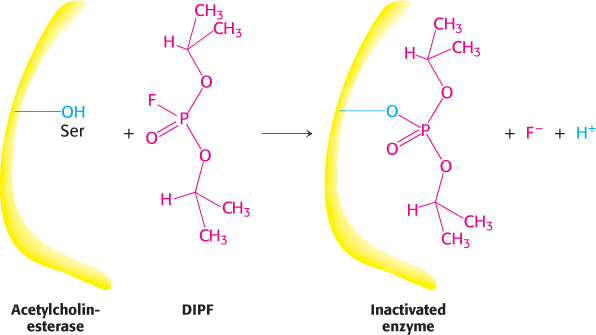
FIGURE 8.22Enzyme inhibition by diisopropylphosphofluoridate (DIPF), a group-specific reagent. DIPF can inhibit an enzyme by covalently modifying a crucial serine residue.
Group-specific reagents react with specific side chains of amino acids. An example of a group-specific reagent is diisopropylphosphofluoridate (DIPF). DIPF modifies only 1 of the 28 serine residues in the proteolytic enzyme chymotrypsin and yet inhibits the enzyme, implying that this serine residue is especially reactive. We will see in Chapter 9 that this serine residue is indeed located at the active site. DIPF also revealed a reactive serine residue in acetylcholinesterase, an enzyme important in the transmission of nerve impulses (Figure 8.22). Thus, DIPF and similar compounds that bind and inactivate acetylcholinesterase are potent nerve gases. Most group-specific reagents do not display the exquisite specificity shown by DIPF. Consequently, more specific means of modifying the active site are required.
Affinity labels, or reactive substrate analogs, are molecules that are structurally similar to the substrate for an enzyme and that covalently bind to active-site residues. They are thus more specific for the enzyme’s active site than are group-specific reagents. Tosyl-l-phenylalanine chloromethyl ketone (TPCK) is a substrate analog for chymotrypsin (Figure 8.23). TPCK binds at the active site and then reacts irreversibly with a histidine residue at that site, inhibiting the enzyme. The compound 3-bromoacetol phosphate is an affinity label for the enzyme triose phosphate isomerase (TPI). It mimics the normal substrate, dihydroxyacetone phosphate, by binding at the active site; then it covalently modifies the enzyme such that the enzyme is irreversibly inhibited (Figure 8.24).
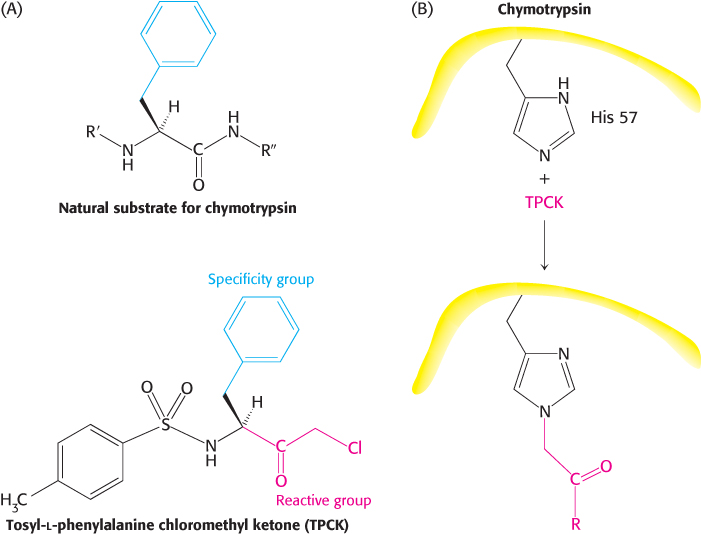
FIGURE 8.23Affinity labeling. (A) Tosyl-l-phenylalanine chloromethyl ketone (TPCK) is a reactive analog of the normal substrate for the enzyme chymotrypsin. (B) TPCK binds at the active site of chymotrypsin and modifies an essential histidine residue.
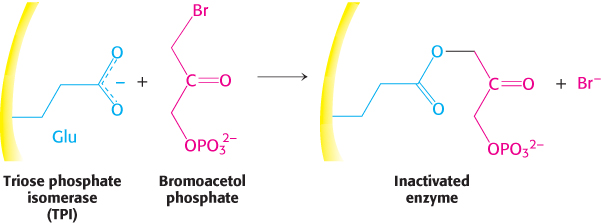
FIGURE 8.24Bromoacetol phosphate, an affinity label for triose phosphate isomerase (TPI). Bromoacetol phosphate, an analog of dihydroxyacetone phosphate, binds at the active site of the enzyme and covalently modifies a glutamic acid residue required for enzyme activity.
Suicide inhibitors, or mechanism-based inhibitors, are modified substrates that provide the most specific means for modifying an enzyme’s active site. The inhibitor binds to the enzyme as a substrate and is initially processed by the normal catalytic mechanism. The mechanism of catalysis then generates a chemically reactive intermediate that inactivates the enzyme through covalent modification. The fact that the enzyme participates in its own irreversible inhibition strongly suggests that the covalently modified group on the enzyme is vital for catalysis. An example is N,N-dimethylpropargylamine, an inhibitor of the enzyme monoamine oxidase (MAO). A flavin prosthetic group of monoamine oxidase oxidizes the N,N-dimethylpropargylamine, which in turn inactivates the enzyme by binding to N-5 of the flavin prosthetic group (Figure 8.25). Monoamine oxidase deaminates neurotransmitters such as dopamine and serotonin, lowering their levels in the brain. Parkinson disease is associated with low levels of dopamine, and depression is associated with low levels of serotonin. N,N-Dimethylpropargylamine and (−)deprenyl, another suicide inhibitor of monoamine oxidase, are used to treat Parkinson disease and depression.
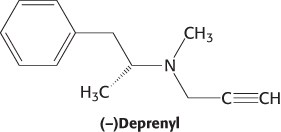
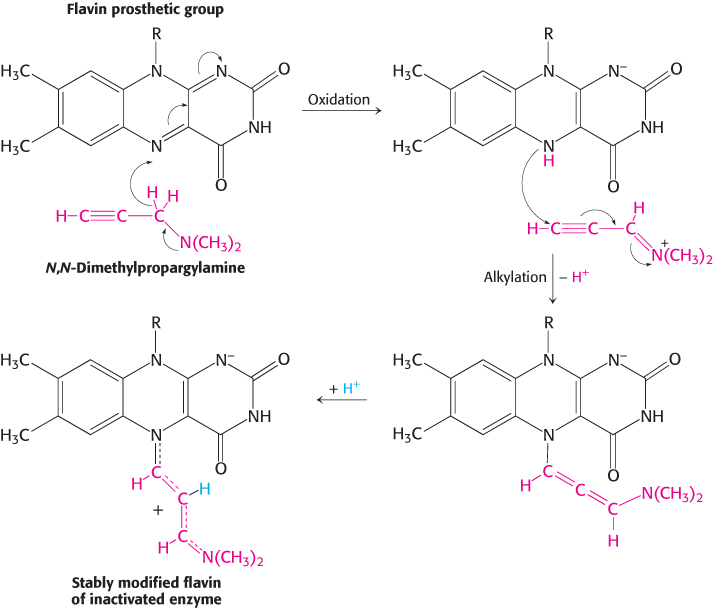
FIGURE 8.25Mechanism-based (suicide) inhibition. Monoamine oxidase, an enzyme important for neurotransmitter synthesis, requires the cofactor FAD (flavin adenine dinucleotide). N,N-Dimethylpropargylamine inhibits monoamine oxidase by covalently modifying the flavin prosthetic group only after the inhibitor has been oxidized. The N-5 flavin adduct is stabilized by the addition of a proton. R represents the remainder of the flavin prosthetic group.
Penicillin irreversibly inactivates a key enzyme in bacterial cell-wall synthesis
 Penicillin, the first antibiotic discovered, provides us with another example of a clinically useful suicide inhibitor. Penicillin consists of a thiazolidine ring fused to a β-lactam ring to which a variable R group is attached by a peptide bond (Figure 8.26A). In benzylpenicillin, for example, R is a benzyl group (Figure 8.26B). This structure can undergo a variety of rearrangements, and, in particular, the β-lactam ring is very labile. Indeed, this instability is closely tied to the antibiotic action of penicillin, as will be evident shortly.
Penicillin, the first antibiotic discovered, provides us with another example of a clinically useful suicide inhibitor. Penicillin consists of a thiazolidine ring fused to a β-lactam ring to which a variable R group is attached by a peptide bond (Figure 8.26A). In benzylpenicillin, for example, R is a benzyl group (Figure 8.26B). This structure can undergo a variety of rearrangements, and, in particular, the β-lactam ring is very labile. Indeed, this instability is closely tied to the antibiotic action of penicillin, as will be evident shortly.
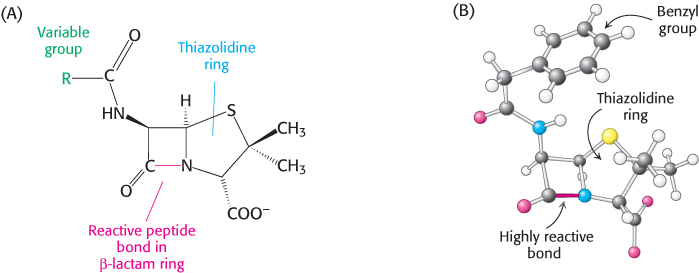
FIGURE 8.26The reactive site of penicillin is the peptide bond of its β-lactam ring. (A) Structural formula of penicillin. (B) Representation of benzylpenicillin.
How does penicillin inhibit bacterial growth? Let us consider Staphylococcus aureus, the most common cause of staph infections. Penicillin interferes with the synthesis of the S. aureus cell wall. The S. aureus cell wall is made up of a macromolecule, called a peptidoglycan (Figure 8.27), which consists of linear polysaccharide chains that are cross-linked by short peptides (pentaglycines and tetrapeptides). The enormous peptidoglycan molecule confers mechanical support and prevents bacteria from bursting in response to their high internal osmotic pressure. Glycopeptide transpeptidase catalyzes the formation of the cross-links that make the peptidoglycan so stable (Figure 8.28). Bacterial cell walls are distinctive in containing D amino acids, which form cross-links by a mechanism different from that used to synthesize proteins.
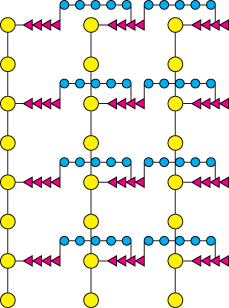
FIGURE 8.27Schematic representation of the peptidoglycan in Staphylococcus aureus. The sugars are shown in yellow, the tetrapeptides in red, and the pentaglycine bridges in blue. The cell wall is a single, enormous, bag-shaped macromolecule because of extensive cross-linking.
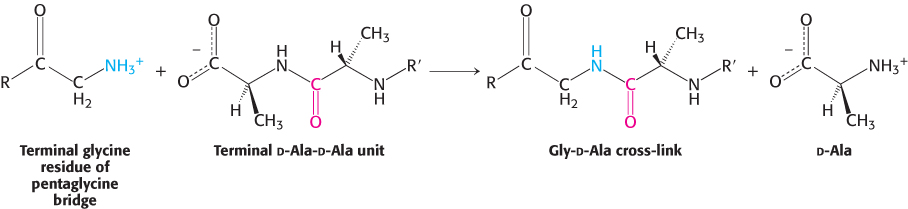
FIGURE 8.28Formation of cross-links in S. aureus peptidoglycan. The terminal amino group of the pentaglycine bridge in the cell wall attacks the peptide bond between two d-alanine residues to form a cross-link.
Penicillin inhibits the cross-linking transpeptidase by the Trojan horse stratagem. The transpeptidase normally forms an acyl intermediate with the penultimate d-alanine residue of the d-Ala-d-Ala peptide (Figure 8.29). This covalent acyl-enzyme intermediate then reacts with the amino group of the terminal glycine in another peptide to form the cross-link. Penicillin is welcomed into the active site of the transpeptidase because it mimics the d-Ala-d-Ala moiety of the normal substrate (Figure 8.30). Bound penicillin then forms a covalent bond with a serine residue at the active site of the enzyme. This penicilloyl-enzyme does not react further. Hence, the transpeptidase is irreversibly inhibited and cell-wall synthesis cannot take place.
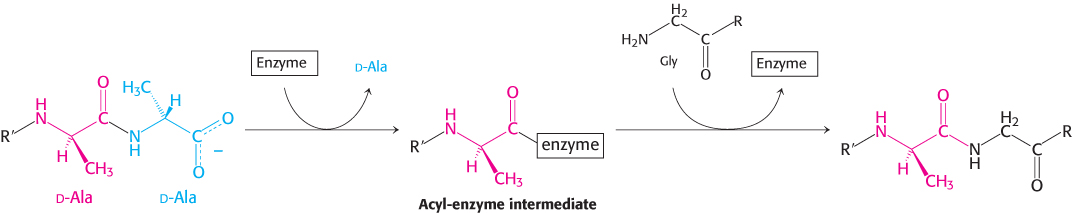
FIGURE 8.29Transpeptidation reaction. An acyl-enzyme intermediate is formed in the transpeptidation reaction leading to cross-link formation.
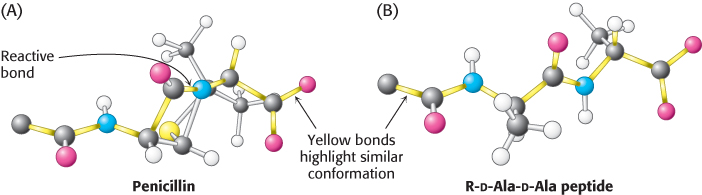
FIGURE 8.30Conformations of penicillin and a normal substrate. The conformation of penicillin in the vicinity of its reactive peptide bond (A) resembles the postulated conformation of the transition state of R-d-Ala-d-Ala (B) in the transpeptidation reaction.
[Information from B. Lee, J. Mol. Biol. 61:463–469, 1971.]
Why is penicillin such an effective inhibitor of the transpeptidase? The highly strained, four-membered β-lactam ring of penicillin makes it especially reactive (Figure 8.26). On binding to the transpeptidase, the serine residue at the active site attacks the carbonyl carbon atom of the lactam ring to form the penicilloyl-serine derivative (Figure 8.31). Because the peptidase participates in its own inactivation, penicillin acts as a suicide inhibitor.
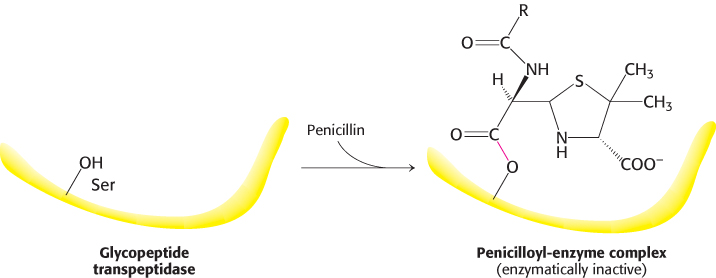
FIGURE 8.31Formation of a penicilloyl-enzyme derivative. Penicillin reacts irreversibly with the transpeptidase to inactivate the enzyme.
Catalytic antibodies demonstrate the importance of selective binding of the transition state to enzymatic activity
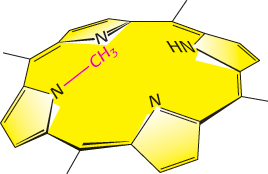
FIGURE 8.33N-Methylmesoporphyrin is a transition-state analog used to generate catalytic antibodies. The insertion of a metal ion into a porphyrin by ferrochelatase proceeds through a transition state in which the porphyrin is bent. N-Methylmesoporphyrin, a bent porphyrin that resembles the transition state of the ferrochelatase-catalyzed reaction, was used to generate an antibody that also catalyzes the insertion of a metal ion into a porphyrin ring.
Recall that antibodies bind precisely to their targets (antigens), and that specific antibodies can be generated against any antigen (Section 3.3). Antibodies that recognize transition states should function as catalysts, if our understanding of the importance of the transition state to catalysis is correct. The preparation of an antibody that catalyzes the insertion of a metal ion into a porphyrin nicely illustrates the validity of this approach. Ferrochelatase, the final enzyme in the biosynthetic pathway for the production of heme, catalyzes the insertion of Fe2+ into protoporphyrin IX (Section 24.4). The nearly planar porphyrin must be bent for iron to enter.
The challenge was to find a transition-state analog for this metallation reaction that could be used as an antigen (immunogen) to generate an antibody. The solution came from studies showing that an alkylated porphyrin, N-methylmesoporphyrin, is a potent inhibitor of ferrochelatase (Figure 8.33). This compound resembles the transition state because N-alkylation forces the porphyrin to be bent. Moreover, N-alkylporphyrins were known to chelate metal ions 104 times as fast as their unalkylated counterparts do. Bending increases the exposure of the pyrrole nitrogen lone pairs of electrons to solvent, which enables the binding of the iron ion.
An antibody catalyst was produced with the use of an N-alkylporphyrin as the antigen. The resulting antibody presumably distorts a planar porphyrin to facilitate the entry of a metal ion. On average, an antibody molecule metallated 80 porphyrin molecules per hour, a rate only 10-fold less than that of ferrochelatase, and 2500-fold faster than the uncatalyzed reaction. Catalytic antibodies (abzymes) can indeed be produced by using transition-state analogs as antigens. Antibodies catalyzing many other kinds of chemical reactions such as ester and amide hydrolysis have been produced by using similar strategies. Studies with transition-state analogs provide strong evidence that enzymes function by assuming a conformation in the active site that is complementary in structure to the transition state. The power of transition-state analogs is now evident: (1) they are sources of insight into catalytic mechanisms, (2) they can serve as potent and specific inhibitors of enzymes, and (3) they can be used as immunogens to generate a wide range of novel catalysts.