26.3
The Complex Regulation of Cholesterol Biosynthesis Takes Place at Several Levels
Cholesterol can be obtained from the diet or it can be synthesized de novo. Cholesterol biosynthesis is one of the most highly regulated metabolic pathways known. Biosynthetic rates may vary several hundredfold, depending on how much cholesterol is consumed in the diet. An adult on a low-cholesterol diet typically synthesizes about 800 mg of cholesterol per day. The liver is the major site of cholesterol synthesis in mammals, although the intestine also forms significant amounts. The rate of cholesterol formation by these organs is highly responsive to the cellular level of cholesterol. This feedback regulation is mediated primarily by changes in the amount and activity of 3-hydroxy-3-methylglutaryl CoA reductase. As described earlier, this enzyme catalyzes the formation of mevalonate, the committed step in cholesterol biosynthesis. HMG CoA reductase is controlled in multiple ways:
1. The rate of synthesis of reductase mRNA is controlled by the sterol regulatory element binding protein (SREBP), a member of a family of transcription factors that regulate the proteins required for lipid synthesis. This transcription factor binds to a short DNA sequence called the sterol regulatory element (SRE) on the 5′ side of the reductase gene when cholesterol levels are low, enhancing transcription of the gene. In its inactive state, the SREBP resides in the endoplasmic reticulum membrane, where it is associated with the SREBP cleavage activating protein (SCAP), an integral membrane protein. SCAP is the cholesterol sensor. When cholesterol levels fall, SCAP escorts SREBP in small membrane vesicles to the Golgi complex, where it is released from the membrane by two specific proteolytic cleavages (Figure 26.14). The first cleavage frees a fragment of SREBP from SCAP, whereas the second cleavage releases the regulatory domain from the membrane. The released protein migrates to the nucleus and binds the SRE of the HMG-CoA reductase gene, as well as several other genes in the cholesterol biosynthetic pathway, to enhance transcription. When cholesterol levels rise, the proteolytic release of the SREBP is blocked, and the SREBP in the nucleus is rapidly degraded by proteasomes located in the nucleus. These two events halt the transcription of genes of the cholesterol biosynthetic pathways.

FIGURE 26.14The SREBP pathway. SREBP resides in the endoplasmic reticulum (ER), where it is bound to SCAP by its regulatory (Reg) domain. When cholesterol levels fall, SCAP and SREBP move to the Golgi complex, where SREBP undergoes successive proteolytic cleavages by a serine protease and a metalloprotease. The released DNA-binding domain moves to the nucleus to alter gene expression.
[Information from an illustration provided by Dr. Michael Brown and Dr. Joseph Goldstein.]
What is the molecular mechanism that retains SCAP–SREBP in the ER when cholesterol is present but allows movement to the Golgi complex when cholesterol concentration is low? When cholesterol is low, SCAP binds to vesicular proteins that facilitate the transport of SCAP–SREBP to the Golgi apparatus, as heretofore described. When cholesterol is present, SCAP binds cholesterol, which causes a structural change in SCAP so that it binds to another endoplasmic reticulum protein called Insig (insulin-induced gene) (Figure 26.15). Insig is the anchor that retains SCAP and thus SREBP in the endoplasmic reticulum in the presence of cholesterol. The interactions between SCAP and Insig can also be forged when Insig binds 25-hydroxycholesterol, a metabolite of cholesterol. Thus, two distinct steroid–protein interactions prevent the inappropriate movement of SCAP–SREBP to the Golgi complex.

FIGURE 26.15Insig regulates SCAP–SREBP movement. In the presence of cholesterol, Insig interacts with SCAP–SREBP and prevents the activation of SREBP. Cholesterol binding to SCAP or 25-hydroxycholesterol binding to Insig facilitates the interaction of Insig and SCAP, retaining SCAP–SREBP in the endoplasmic reticulum.
[Information from M. S. Brown and J. L. Goldstein. Cholesterol feedback: From Schoenheimer’s bottle to Scap’s MELADL, J. Lipid Res. 50:S15–S27, 2009.]
2. The rate of translation of reductase mRNA is inhibited by nonsterol metabolites derived from mevalonate.
3. The degradation of the reductase is stringently controlled. The enzyme is bipartite: its cytoplasmic domain carries out catalysis and its membrane domain senses signals that lead to its degradation. The membrane domain may undergo structural changes in response to increasing concentrations of sterols such as lanosterol and 25-hydroxycholesterol. Under these conditions, the reductase appears to bind to a subset of Insigs that are also associated with the ubiquitinating enzymes (Figure 26.16). The reductase is polyubiquitinated and subsequently extracted from the membrane in a process that requires gerenylgeraniol. The extracted reductase is then degraded by the proteasome (Section 23.2). The combined regulation at the levels of transcription, translation, and degradation can alter the amount of enzyme in the cell more than 200-fold.
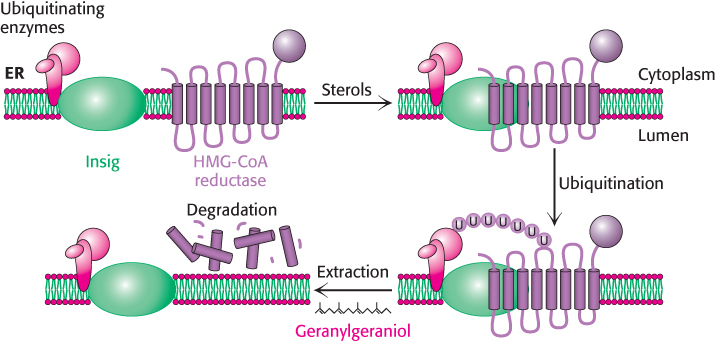
FIGURE 26.16Insig facilitates the degradation of HMG-CoA reductase. In the presence of sterols, a subclass of Insig associated with ubiquitinating enzymes binds HMG-CoA reductase. This interaction results in the ubiquitination of the enzyme. This modification and the presence of geranylgeraniol results in extraction of the enzyme from the membrane and degradation by the proteasome.
[Information from R. A. DeBose-Boyd. Feedback regulation of cholesterol synthesis: Sterol-accelerated ubiquitination and degradation of HMG CoA reductase, Cell Res. 18:609–621, 2008.]
4. Phosphorylation decreases the activity of the reductase. This enzyme, like acetyl CoA carboxylase (which catalyzes the committed step in fatty acid synthesis, Section 22.5), is switched off by an AMP-activated protein kinase. Thus, cholesterol synthesis ceases when the ATP level is low.
Lipoproteins transport cholesterol and triacylglycerols throughout the organism
Cholesterol and triacylglycerols are transported in body fluids in the form of lipoprotein particles, which are important for a number of reasons. First, lipoprotein particles are the means by which triacylglycerols are delivered to tissues, from the intestine or liver, for use as fuel or for storage. Second, the fatty acid constituents of the triacylglycerol components of the lipoprotein particles are incorporated into phospholipids for membrane synthesis. Likewise, cholesterol is a vital component of membranes and is a precursor to the powerful signal molecules, the steroid hormones. Finally, cells are not able to degrade the steroid nucleus. Consequently, the cholesterol must be used biochemically or excreted by the liver. Excess cholesterol plays a role in the development of atherosclerosis. Lipoprotein particles function in cholesterol homeostasis, transporting the molecule from sites of synthesis to sites of use, and finally to the liver for excretion.
Each lipoprotein particle consists of a core of hydrophobic lipids surrounded by a shell of more-polar lipids and proteins. The protein components of these macromolecular aggregates, called apoproteins, have two roles: they solubilize hydrophobic lipids and contain cell-targeting signals. Apolipoproteins are synthesized and secreted by the liver and the intestine. Lipoprotein particles are classified according to increasing density (Table 26.1): chylomicrons, chylomicron remnants, very low density lipoproteins (VLDLs), intermediate-density lipoproteins (IDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs). These classes have numerous subtypes. Moreover, lipoprotein particles can shift between classes as they release or pick up cargo, thereby changing their density.
TABLE 26.1 Properties of plasma lipoproteins
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Very low density lipoprotein |
|
|
|
|
|
|
|
|
|
Intermediate-density lipoprotein |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Reverse cholesterol transport |
|
|
|
|
|
Abbreviations: TAG, triacylglyerol; CE, cholesteryl ester; C, free cholesterol; PL, phospholipid; P, protein. |
Triacylglycerols, cholesterol, and other lipids obtained from the diet are carried away from the intestine in the form of large chylomicrons (Section 22.1). These particles have a very low density because triacylglycerols constitute about 90% of their content. Apolipoprotein B-48 (apo B-48), a large protein (240 kDa), forms an amphipathic spherical shell around the fat globule; the external face of this shell is hydrophilic. The triacylglycerols in chylomicrons are released through hydrolysis by lipoprotein lipases. These enzymes are located on the lining of blood vessels in muscle and other tissues that use fatty acids as fuels or in the synthesis of lipids. The liver then takes up the cholesterol-rich residues, known as chylomicron remnants.
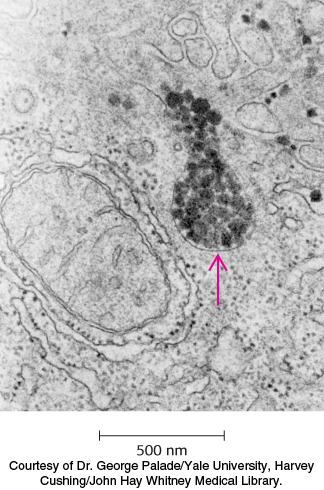
FIGURE 26.17Site of cholesterol synthesis. Electron micrograph of a part of a liver cell actively engaged in the synthesis and secretion of very low density lipoprotein (VLDL). The arrow points to a vesicle that is releasing its content of VLDL particles.
Lipoprotein particles are also crucial for the transport of lipids from the liver, which is a major site of triacylglycerol and cholesterol synthesis, to other tissues in the body (Figure 26.17). Triacylglycerols and cholesterol in excess of the liver’s own needs are exported into the blood in the form of very low density lipoproteins. These particles are stabilized by two apolipoproteins—apo B-100 (513 kDa) and apo E (34 kDa). Triacylglycerols in very low density lipoproteins, as in chylomicrons, are hydrolyzed by lipases on capillary surfaces, with the released fatty acids being taken up by the muscle and other tissues. The resulting remnants, which are rich in cholesteryl esters, are called intermediate-density lipoproteins. These particles have two fates. Half of them are taken up by the liver for processing, and half are converted into low-density lipoprotein by the removal of more triacylglycerol by tissue lipases that absorb the released fatty acids. Interestingly, apo B-100, one of the largest proteins known, is a longer version of apo B-48, the protein component of chylomicrons. Both apo B proteins are encoded by the same gene and produced from the same initial RNA transcript. In the intestine, RNA editing (Section 29.3) modifies the transcript to generate the mRNA for apo B-48, the truncated form.
Low-density lipoprotein is the major carrier of cholesterol in blood (Figure 26.18). It contains a core of some 1500 cholesterol molecules esterified to fatty acids; the most common fatty acid chain in these esters is linoleate, a polyunsaturated fatty acid. A shell of phospholipids and unesterified cholesterol molecules surrounds this highly hydrophobic core. The shell also contains a single copy of apo B-100, which is recognized by target cells. The role of LDL is to transport cholesterol to peripheral tissues and regulate de novo cholesterol synthesis at these sites. A different purpose is served by high-density lipoprotein, which picks up cholesterol released into the plasma from dying cells and from membranes undergoing turnover and delivers the cholesterol to the liver for excretion. An acyltransferase in HDL esterifies these cholesterols, which are then returned by HDL to the liver (Figure 26.19).

FIGURE 26.18Schematic model of low-density lipoprotein. The LDL particle is approximately 22 nm (220 Å) in diameter.
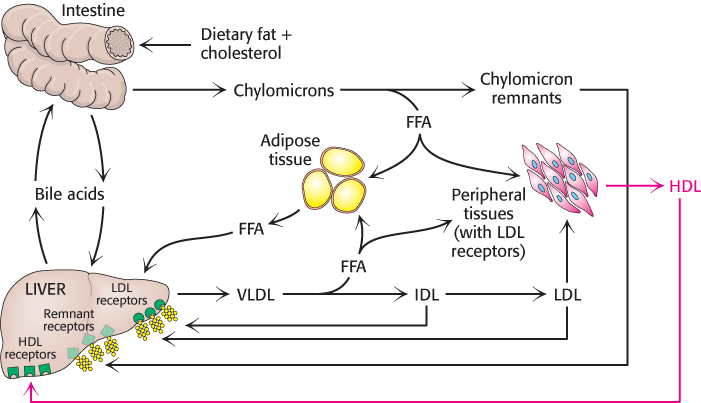
FIGURE 26.19An overview of lipoprotein particle metabolism. Fatty acids are abbreviated FFA.
[Information from J. G. Hardman (Ed.), L. L. Limbird (Ed.), and A. G. Gilman (Consult. Ed.), Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 10th ed. (McGraw-Hill, 2001), p. 975, Fig. 36.1.]
Low-density lipoproteins play a central role in cholesterol metabolism
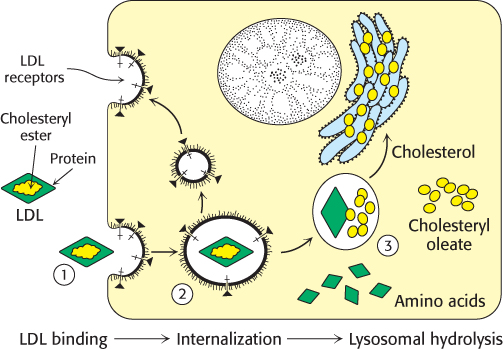
FIGURE 26.20Receptor-mediated endocytosis. The process of receptor-mediated endocytosis is illustrated for the cholesterol-carrying complex, low-density lipoprotein (LDL): (1) LDL binds to a specific receptor, the LDL receptor; (2) this complex invaginates to form an endosome; (3) after separation from its receptor, the LDL-containing vesicle fuses with a lysosome, leading to the degradation of the LDL and the release of the cholesterol.
Cholesterol metabolism must be precisely regulated to prevent atherosclerosis. The mode of control in the liver, the primary site of cholesterol synthesis, has already been considered: dietary cholesterol reduces the activity and amount of 3-hydroxy-3-methylglutaryl CoA reductase, the enzyme catalyzing the committed step. In general, cells outside the liver and intestine obtain cholesterol from the plasma rather than synthesizing it de novo. Specifically, their primary source of cholesterol is the low-density lipoprotein. The process of LDL uptake, called receptor-mediated endocytosis, serves as a paradigm for the uptake of many molecules (Figure 26.20).
Endocytosis begins when apolipoprotein B-100 on the surface of an LDL particle binds to a specific receptor protein on the plasma membrane of nonhepatic cells. The receptors for LDL are localized in specific regions called coated pits, which contain a protein called clathrin. The receptor–LDL complex is then internalized by endocytosis; that is, the plasma membrane in the vicinity of the complex invaginates and then fuses to form an endocytic vesicle called an endosome. The endosome is acidified by an ATP-dependent proton pump homologous to the Na+/K+ ATPase (Section 13.2), which causes the receptor to release its cargo. Some of the receptor is returned to the cell membrane in a recycling vesicle, while a portion is degraded along with its cargo. The round-trip time for a receptor is about 10 minutes; in its lifetime of about a day, each receptor may bring hundreds of LDL particles into the cell. The vesicles containing LDL and in some cases the receptor subsequently fuse with lysosomes, acidic vesicles that carry a wide array of degradative enzymes. The protein component of LDL is hydrolyzed to free amino acids. The cholesteryl esters in LDL are hydrolyzed by a lysosomal acid lipase. The released unesterified cholesterol can then be used for membrane biosynthesis. Alternatively, it can be reesterified for storage inside the cell. In fact, free cholesterol activates acyl CoA:cholesterol acyltransferase (ACAT), the enzyme catalyzing this reaction. Reesterified cholesterol contains mainly oleate and palmitoleate, which are monounsaturated fatty acids, in contrast with the cholesteryl esters in LDL, which are rich in linoleate, a polyunsaturated fatty acid (Table 12.1). It is imperative that the cholesterol be reesterified. High concentrations of unesterified cholesterol disrupt the integrity of cell membranes.
The synthesis of the LDL receptor is itself subject to feedback regulation. Studies of cultured fibroblasts show that, when cholesterol is abundant inside the cell, new LDL receptors are not synthesized, and so the uptake of additional cholesterol from plasma LDL is blocked. The gene for the LDL receptor, like that for the reductase, is regulated by SREBP, which binds to a sterol regulatory element that controls the rate of mRNA synthesis.
The absence of the LDL receptor leads to hypercholesterolemia and atherosclerosis
 The pioneering studies of familial hypercholesterolemia by Michael Brown and Joseph Goldstein revealed the physiological importance of the LDL receptor. The total concentration of cholesterol and LDL in the blood plasma is markedly elevated in this genetic disorder, which results from a mutation at a single autosomal locus. The cholesterol level in the plasma of homozygotes is typically 680 mg dl−1, compared with 300 mg dl−1 in heterozygotes (clinical assay results are often expressed in milligrams per deciliter, which is equal to milligrams per 100 milliliters). A value of < 200 mg dl−1 is regarded as desirable, but many people have higher levels. In familial hypercholesterolemia, cholesterol is deposited in various tissues because of the high concentration of LDL cholesterol in the plasma. Nodules of cholesterol called xanthomas are prominent in skin and tendons. LDL accumulates under the endothelial cells lining the blood vessels. Of particular concern is the oxidation of the excess LDL to form oxidized LDL (oxLDL), which can instigate the inflammatory response by the immune system, a response that has been implicated in the development of cardiovascular disease. The oxLDL is taken up by immune system cells called macrophages, which become engorged to form foam cells. These foam cells become trapped in the walls of the blood vessels and contribute to the formation of atherosclerotic plaques that cause arterial narrowing and lead to heart attacks (Figure 26.21). In fact, most homozygotes die of coronary artery disease in childhood. The disease in heterozygotes (1 in 500 people) has a milder and more variable clinical course.
The pioneering studies of familial hypercholesterolemia by Michael Brown and Joseph Goldstein revealed the physiological importance of the LDL receptor. The total concentration of cholesterol and LDL in the blood plasma is markedly elevated in this genetic disorder, which results from a mutation at a single autosomal locus. The cholesterol level in the plasma of homozygotes is typically 680 mg dl−1, compared with 300 mg dl−1 in heterozygotes (clinical assay results are often expressed in milligrams per deciliter, which is equal to milligrams per 100 milliliters). A value of < 200 mg dl−1 is regarded as desirable, but many people have higher levels. In familial hypercholesterolemia, cholesterol is deposited in various tissues because of the high concentration of LDL cholesterol in the plasma. Nodules of cholesterol called xanthomas are prominent in skin and tendons. LDL accumulates under the endothelial cells lining the blood vessels. Of particular concern is the oxidation of the excess LDL to form oxidized LDL (oxLDL), which can instigate the inflammatory response by the immune system, a response that has been implicated in the development of cardiovascular disease. The oxLDL is taken up by immune system cells called macrophages, which become engorged to form foam cells. These foam cells become trapped in the walls of the blood vessels and contribute to the formation of atherosclerotic plaques that cause arterial narrowing and lead to heart attacks (Figure 26.21). In fact, most homozygotes die of coronary artery disease in childhood. The disease in heterozygotes (1 in 500 people) has a milder and more variable clinical course.

FIGURE 26.21The effects of excess cholesterol. Cross section of (A) a normal artery and (B) an artery blocked by a cholesterol-rich plaque.
The molecular defect in most cases of familial hypercholesterolemia is an absence or deficiency of functional receptors for LDL. Receptor mutations that disrupt each of the stages in the endocytotic pathway have been identified. Homozygotes have almost no functional receptors for LDL, whereas heterozygotes have about half the normal number. Consequently, the entry of LDL into liver and other cells is impaired, leading to an increased level of LDL in the blood plasma. Furthermore, less IDL enters liver cells because IDL entry, too, is mediated by the LDL receptor. Consequently, IDL stays in the blood longer in familial hypercholesterolemia, and more of it is converted into LDL than in normal people. All deleterious consequences of an absence or deficiency of the LDL receptor can be attributed to the ensuing elevated level of LDL cholesterol in the blood.
Mutations in the LDL receptor prevent LDL release and result in receptor destruction
One class of mutations that results in familial hypercholesterolemia generates receptors that are reluctant to give up the LDL cargo. Let us begin by examining the makeup of the receptor. The human LDL receptor is a 160-kDa glycoprotein, which is composed of six different types of domains (Figure 26.22A). The amino-terminal region of the receptor, which is the site of LDL binding, consists of seven homologous LDL receptor type-A (LA) domains, with domains 4 and 5 most important for LDL binding. A second type of domain is homologous to one found in the epidermal growth factor (EGF). This domain is repeated three times, and in between the second and third repeat is a propeller structure consisting of six bladelike domains. This part of the receptor is crucial in releasing LDL in the endosome. The fourth domain, which is very rich in serine and threonine residues, contains O-linked sugars. These oligosaccharides function as struts to keep the receptor extended from the membrane so that the LDL-binding domain is accessible to LDL. The fifth type of domain consists of 22 hydrophobic residues that span the plasma membrane. The sixth and final domain consists of 50 residues and emerges on the cytoplasmic side of the membrane, where it controls the interaction of the receptor with coated pits and participates in endocytosis.

FIGURE 26.22LDL receptor releases LDL in the endosome. (A) A schematic representation of the domain structure of the LDL receptor. (B) In the endosome, the receptor converts from the open structure into the closed structure, resulting in the release of the LDL into the endosome.
[Information from I. D. Campbell, Biochem. Soc. Trans. 31 (pt. 6p):1107–1114, 2003, Fig. 1A.]
How does the LDL receptor relinquish its cargo on entering the endosome? The receptor exists in two interconvertible states: an extended or open state, capable of binding LDL, and a closed state that results in release of the LDL in the endosome. The receptor maintains the open state while in the plasma membrane, on binding LDL, and throughout its journey to the endosome. The conversion from the open state into the closed state takes place on exposure to the acidic environment of the endosome (Figure 26.22B). Three contiguous modules LA7, EGFA, and EGFB rigidly position the propeller module to facilitate displacement of the LDL as the closed state is formed. Under neutral pH, aspartate residues of the propeller blades form hydrogen bonds that hold each blade to the rest of the propeller structure. Exposure to the low-pH environment of the endosome causes the propeller-like structures to interact with the LDL-binding domain. This interaction displaces the LDL, which is then digested by the lysosome. The receptor is often returned to the plasma membrane to again bind LDL. The importance of this process is highlighted by the fact that more than half of the point mutations that result in familial hypercholesterolemia are due to disruptions in the interconversion between the open state and the closed state. These mutations result in a failure to release the LDL cargo and loss of the receptor by degradation.
Cycling of the LDL receptor is regulated
PCSK9 (proprotein convertase subtilisin/kexin type) is a protease, secreted by the liver, which plays a crucial role in the regulation of cycling of the LDL receptor. Despite the fact that PCSK9 is a protease, enzymatic activity of the protein is not required for cycling regulation. PCSK9 in the blood binds to the EGFA domain of the receptor (Figure 26.22). PCSK9 locks the receptor in the open conformation even in the acidic conditions of the endosome. Failure to adopt the closed conformation prevents the receptor from returning to the plasma membrane, and it is degraded in the lysosome along with its cargo.
Individuals having a mutation that reduces the amount of PCSK9 in the blood have greatly reduced levels of LDL in the blood and display an almost 90% reduction in the rate of cardiovascular disease. Presumably, reduced levels of PCSK9 allow more receptor cycling and more efficient removal of LDL from the blood. Much research in now being directed at inhibiting PCSK9-mediated receptor degradation in individuals with high cholesterol levels. Phase 3 trials (Section 36.4) are underway, testing the efficacy of a monoclonal antibody to PCSK9. Monoclonal antibody treatments are expensive, so the search continues for a chemical inhibitor of the protein.
HDL appears to protect against atherosclerosis
Although the events that result in atherosclerosis take place rapidly in familial hypercholesterolemia, a similar sequence of events takes place in people who develop atherosclerosis over decades. In particular, the formation of foam cells and plaques are especially hazardous occurrences. HDL and its role in returning cholesterol to the liver appear to be important in mitigating these life-threatening circumstances.
HDL has a number of antiatherogenic properties, including the inhibition of LDL oxidation, but the best-characterized property is the removal of cholesterol from cells, especially macrophages. Earlier, we learned that HDL retrieves cholesterol from other tissues in the body to return the cholesterol to the liver for excretion as bile or in the feces. This transport, called reverse cholesterol transport, is especially important in regard to macrophages. Indeed, when the transport fails, macrophages become foam cells and facilitate the formation of plaques. Macrophages that collect cholesterol from LDL normally transport the cholesterol to HDL particles. The more HDL, the more readily this transport takes place and the less likely that the macrophages will develop into foam cells. Presumably, this robust reverse cholesterol transport accounts for the observation that higher HDL levels confer protection against atherosclerosis.
The importance of reverse cholesterol transport is illustrated by the occurrence of mutations that inactivate a cholesterol-transport protein in endothelial cells and macrophages, ABCA1 (ATP-binding cassette transporter, subfamily A1) (Figure 13.7). Loss of activity of cholesterol-transport protein ABCA1 results in a very rare condition called Tangier disease, which is characterized by HDL deficiency, accumulation of cholesterol in macrophages, and premature atherosclerosis. Under normal conditions, the apoprotein component of HDL, apoA-I, binds to ABCA1 to facilitate LDL transport. Moreover, the interaction between apoA-I and ABCA1 initiates a signal transduction pathway in the endothelial cells that inhibits the inflammatory response.
Until recently, high levels of HDL-bound cholesterol (“good cholesterol”) relative to LDL-bound cholesterol (“bad cholesterol”) were believed to protect against cardiovascular disease. This belief was based on epidemiological studies. However, a number of recent clinical trials revealed that increased levels of HDL-bound cholesterol had no protective effects at all. These studies do not discount the protective effects of HDL alone, but they illustrate the danger of equating free HDL and cholesterol-bound HDL.
The clinical management of cholesterol levels can be understood at a biochemical level
Homozygous familial hypercholesterolemia can be treated only by a liver transplant. A more generally applicable therapy is available for heterozygotes and others with high levels of cholesterol. The goal is to reduce the amount of cholesterol in the blood by stimulating the synthesis of more than the customary number of LDL receptors. We have already observed that the production of LDL receptors is controlled by the cell’s need for cholesterol. The therapeutic strategy is to deprive the cell of ready sources of cholesterol. When cholesterol is required, the amount of mRNA for the LDL receptor rises and more receptors are found on the cell surface. This state can be induced by a two-pronged approach. First, the reabsorption of bile salts from the intestine is inhibited. Bile salts are cholesterol derivatives that promote the absorption of dietary cholesterol and dietary fats. Second, de novo synthesis of cholesterol is blocked.

FIGURE 26.23Lovastatin, a competitive inhibitor of HMG-CoA reductase. The part of the structure that resembles the 3-hydroxy-3-methylglutaryl moiety is shown in red.
The reabsorption of bile is impeded by oral administration of positively charged polymers, such as cholestyramine, that bind negatively charged bile salts and are not themselves absorbed. Cholesterol synthesis can be effectively blocked by a class of compounds called statins. A well-known example of such a compound is lovastatin, which is also called mevacor (Figure 26.23). These compounds are potent competitive inhibitors (Ki = 1 nM) of HMG-CoA reductase, the essential control point in the biosynthetic pathway. Plasma cholesterol levels decrease by 50% in many patients given both lovastatin and inhibitors of bile-salt reabsorption. Lovastatin and other inhibitors of HMG-CoA reductase are widely used to lower the plasma-cholesterol level in people who have atherosclerosis, which is the leading cause of death in industrialized societies. Preliminary studies suggest that reducing levels of PCSK9 and HMG-CoA reductase activity may be an especially effective means of reducing cholesterol levels. The development of statins as effective drugs is further described in Chapter 36.