29.1
RNA Polymerases Catalyze Transcription
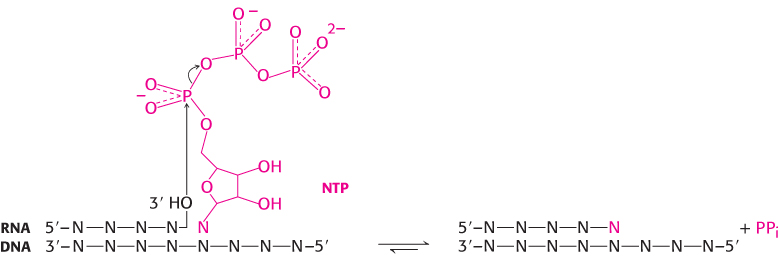
The fundamental reaction of RNA synthesis is the formation of a phosphodiester bond. The 3′-hydroxyl group of the last nucleotide in the chain nucleophilically attacks the α phosphoryl group of the incoming nucleoside triphosphate with the concomitant release of a pyrophosphate.
This reaction is thermodynamically favorable, and the subsequent degradation of the pyrophosphate to orthophosphate locks the reaction in the direction of RNA synthesis. The catalytic sites of RNA polymerases include two metal ions, normally magnesium ions (Figure 29.2). One ion remains tightly bound to the enzyme, whereas the other ion comes in with the nucleoside triphosphate and leaves with the pyrophosphate. Three conserved aspartate residues participate in binding these metal ions.
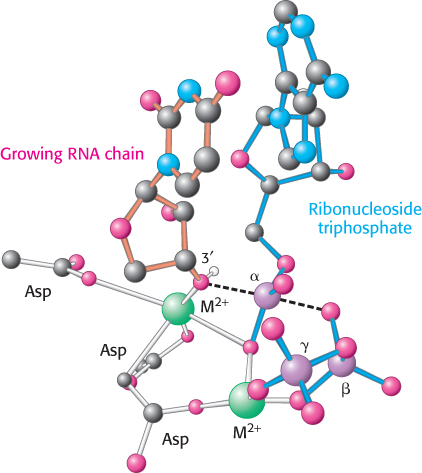
FIGURE 29.2RNA polymerase active site. A model of the transition state for phosphodiesterbond formation in the active site of RNA polymerase. The 3′-hydroxyl group of the growing RNA chain attacks the α-phosphoryl group of the incoming nucleoside triphosphate, resulting in the release of pyrophosphate. This transition state is structurally similar to that in the active site of DNA polymerase (Figure 28.5).
RNA polymerases are very large, complex enzymes. For example, the core of the RNA polymerase of E. coli consists of 5 kinds of subunits with the composition α2ββ′ ω (Table 29.1). A typical eukaryotic RNA polymerase is larger and more complex, having 12 subunits and a total molecular mass of more than 0.5 million daltons. Despite this complexity, the structures of RNA polymerases have been determined in detail by x-ray crystallography in work pioneered by Roger Kornberg and Seth Darst.
TABLE 29.1 Subunits of RNA polymerase from E. coli
The polymerization reactions catalyzed by RNA polymerases take place within a complex in DNA termed a transcription bubble (Figure 29.3). This complex consists of double-stranded DNA that has been locally unwound in a region of approximately 17 base pairs. The edges of the bases that normally take part in Watson–Crick base pairs are exposed in the unwound region. We will begin with a detailed examination of the elongation process, including the role of the DNA template read by RNA polymerase and the reactions catalyzed by the polymerase, before returning to the more-complex processes of initiation and termination.
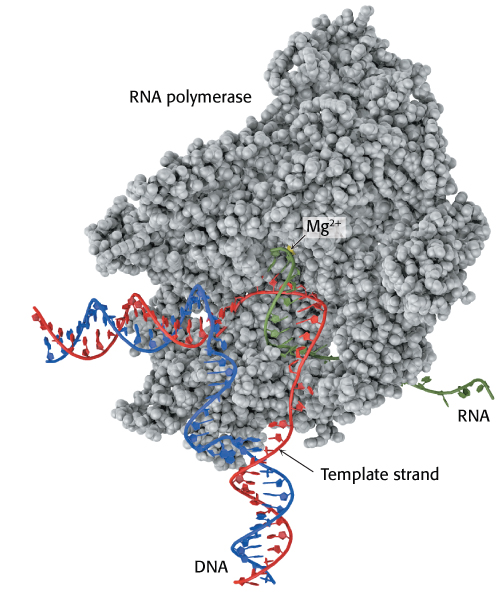
FIGURE 29.3Transcription bubble. RNA polymerase separates a region of the double helix to form a structure called the “transcription bubble.” The red (template) and blue (nontemplate) strands of DNA are shown along with the RNA molecule being synthesized (shown in green). The position of the active site magnesium is indicated. The DNA enters from the left and exits at the bottom.
RNA chains are formed de novo and grow in the 5′-to-3′ direction
Let us begin our examination of transcription by considering the DNA template. The first nucleotide (the start site) of a DNA sequence to be transcribed is denoted as +1 and the second one as +2; the nucleotide preceding the start site is denoted as −1. These designations refer to the coding strand of DNA. Recall that the sequence of the template strand of DNA is the complement of that of the RNA transcript (Figure 29.4). In contrast, the coding strand of DNA has the same sequence as that of the RNA transcript except for thymine (T) in place of uracil (U). The coding strand is also known as the sense (+) strand, and the template strand as the antisense (−) strand.

FIGURE 29.4Template and coding strands. The template, or antisense (−), strand is complementary in sequence to the RNA transcript.
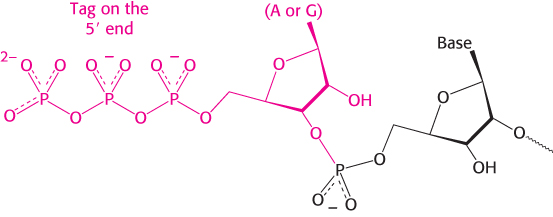
In contrast with DNA synthesis, RNA synthesis can start de novo, without the requirement for a primer. Most newly synthesized RNA chains carry a highly distinctive tag on the 5′ end: the first base at that end is either pppG or pppA.
The presence of the triphosphate moiety confirms that RNA synthesis starts at the 5′ end.
The dinucleotide shown above is synthesized by RNA polymerase as part of the complex process of initiation, which will be discussed later in the chapter. After initiation takes place, RNA polymerase elongates the nucleic acid chain in the following manner (Figure 29.5). A ribonucleoside triphosphate binds in the active site of the RNA polymerase, directly adjacent to the growing RNA chain. The incoming ribonucleoside triphosphate forms a Watson–Crick base pair with the template strand. The 3′-hydroxyl group of the growing RNA chain, oriented and activated by the tightly bound metal ion, attacks the α-phosphoryl group to form a new phosphodiester bond, displacing pyrophosphate.
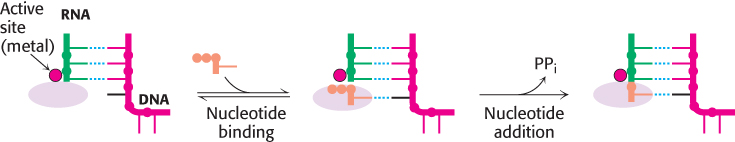
FIGURE 29.5Elongation mechanism. A ribonucleoside triphosphate binds adjacent to the growing RNA chain and forms a Watson–Crick base pair with a base on the DNA template strand. The 3′-hydroxyl group at the end of the RNA chain attacks the newly bound nucleotide and forms a new phosphodiester bond, releasing pyrophosphate.
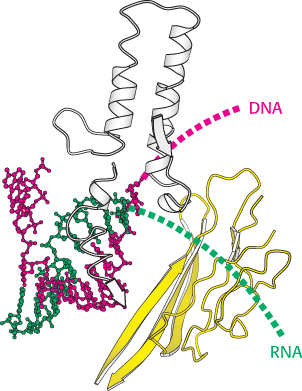
 FIGURE 29.7 RNA–DNA hybrid separation. A structure within RNA polymerase forces the separation of the RNA–DNA hybrid. Notice that the DNA strand exits in one direction and the RNA product exits in another.
FIGURE 29.7 RNA–DNA hybrid separation. A structure within RNA polymerase forces the separation of the RNA–DNA hybrid. Notice that the DNA strand exits in one direction and the RNA product exits in another.
[Drawn from 1I6H.pdb.]
To proceed to the next step, the RNA–DNA hybrid must move relative to the polymerase to bring the 3′ end of the newly added nucleotide into proper position for the next nucleotide to be added (Figure 29.6). This translocation step does not include breaking any bonds between base pairs and is reversible but, once it has taken place, the addition of the next nucleotide, favored by the triphosphate cleavage and pyrophosphate release and cleavage, drives the polymerization reaction forward.
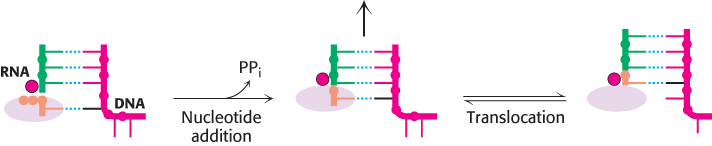
FIGURE 29.6Translocation. After nucleotide addition, the RNA–DNA hybrid can translocate through the RNA polymerase, bringing a new DNA base into position to base-pair with an incoming nucleoside triphosphate.
The lengths of the RNA–DNA hybrid and of the unwound region of DNA stay rather constant as RNA polymerase moves along the DNA template. The length of the RNA–DNA hybrid is determined by a structure within the enzyme that forces the RNA–DNA hybrid to separate, allowing the RNA chain to exit from the enzyme and the DNA chain to rejoin its DNA partner (Figure 29.7).
RNA polymerases backtrack and correct errors
The RNA–DNA hybrid can also move in the direction opposite that of elongation (Figure 29.8). This backtracking is less favorable energetically than moving forward because it breaks the bonds between a base pair. However, backtracking is very important for proofreading. The incorporation of an incorrect nucleotide introduces a non-Watson–Crick base pair. In this case, breaking the bonds between this base pair and backtracking is less costly energetically. After the polymerase has backtracked, the phosphodiester bond one base pair before the one that has just formed is adjacent to the metal ion in the active site. In this position, a hydrolysis reaction in which a water molecule attacks the phosphate can result in the cleavage of the phosphodiester bond and the release of a dinucleotide that includes the incorrect nucleotide.
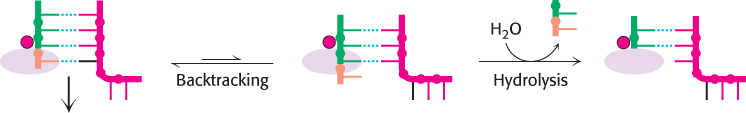
FIGURE 29.8Backtracking. The RNA–DNA hybrid can occasionally backtrack within the RNA polymerase. In the backtracked position, hydrolysis can take place, producing a configuration equivalent to that after translocation. Backtracking is more likely if a mismatched base is added, facilitating proofreading.
Studies of single molecules of RNA polymerase have confirmed that the enzymes hesitate and backtrack to correct errors. Furthermore, these proofreading activities are often enhanced by accessory proteins. The final error rate of the order of one mistake per 104 or 105 nucleotides is higher than that for DNA replication, including all error-correcting mechanisms. The lower fidelity of RNA synthesis can be tolerated because mistakes are not transmitted to progeny. For most genes, many RNA transcripts are synthesized; a few defective transcripts are unlikely to be harmful.
RNA polymerase binds to promoter sites on the DNA template to initiate transcription
The elongation process is common to all organisms. In contrast, the processes of initiation and termination differ substantially in bacteria and eukaryotes. We begin with a discussion of these processes in bacteria, starting with initiation of transcription. The bacterial RNA polymerase discussed earlier with the composition α2ββ′ ω is referred to as the core enzyme. The inclusion of an additional subunit produces the holoenzyme with composition α2ββ′ ωσ. The σ subunit helps find sites on DNA where transcription begins, referred to as promoter sites or, simply, promoters. At these sites, the σ subunit participates in the initiation of RNA synthesis and then dissociates from the rest of the enzyme.
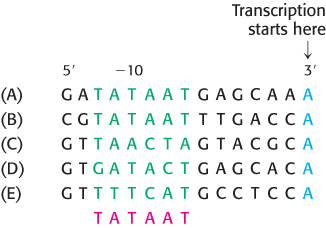
FIGURE 29.9Bacterial promoter sequences. A comparison of five sequences from prokaryotic promoters reveals a recurring sequence of TATAAT centered on position −10. The −10 consensus sequence (in red) was deduced from a large number of promoter sequences. The sequences are from the (A) uvrD, (B) uncI, and (C) trp operons of E. coli, from (D) λ phage, and from (E) φX174 phage.
Sequences upstream of the promoter site are important in determining where transcription begins. A striking pattern was evident when the sequences of bacterial promoters were compared. Two common motifs are present on the upstream side of the transcription start site. They are known as the −10 sequence and the −35 sequence because they are centered at about 10 and 35 nucleotides upstream of the start site. The region containing these sequences is called the core promoter. The −10 and −35 sequences are each 6 bp long. Their consensus sequences, deduced from analyses of many promoters (Figure 29.9), are

Promoters differ markedly in their efficacy. Some genes are transcribed frequently—as often as every 2 seconds in E. coli. The promoters for these genes are referred to as strong promoters. In contrast, other genes are transcribed much less frequently, about once in 10 minutes; the promoters for these genes are weak promoters. The −10 and −35 regions of most strong promoters have sequences that correspond closely to the consensus sequences, whereas weak promoters tend to have multiple substitutions at these sites. Indeed, mutation of a single base in either the −10 sequence or the −35 sequence can diminish promoter activity. The distance between these conserved sequences also is important; a separation of 17 nucleotides is optimal. Thus, the efficiency or strength of a promoter sequence serves to regulate transcription. Regulatory proteins that bind to specific sequences near promoter sites and interact with RNA polymerase (Chapter 31) also markedly influence the frequency of transcription of many genes.
Outside the core promoter in a subset of highly expressed genes is the upstream element (also called the UP element for upstream element). This sequence is present from 40 to 60 nucleotides upstream of the transcription start site. The UP element is bound by the α subunit of RNA polymerase and serves to increase the efficiency of transcription by creating an additional interaction site for the polymerase.
Sigma subunits of RNA polymerase recognize promoter sites
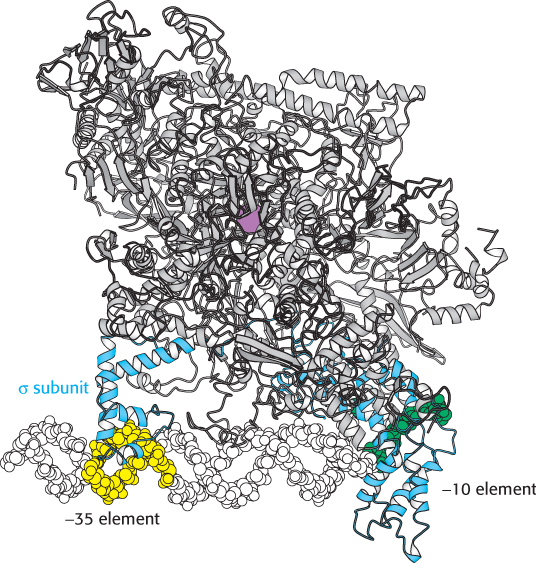
FIGURE 29.10 RNA polymerase holoenzyme complex. Notice that the σ subunit (blue) of the bacterial RNA polymerase holoenzyme makes sequence-specific contacts with the −10 (green) and −35 promoter elements (yellow). The active site of the polymerase is revealed by bound metal ion (purple).
[Drawn from 1L9Z.pdb].
To initiate transcription, the α2ββ′ ω core of RNA polymerase must bind the promoter. However, it is the σ subunit that makes this binding possible by enabling RNA polymerase to recognize promoter sites. In the presence of the σ subunit, the RNA polymerase binds weakly to the DNA and slides along the double helix until it dissociates or encounters a promoter. The σ subunit recognizes the promoter through several interactions with the nucleotide bases of the promoter DNA. The structure of a bacterial RNA polymerase holoenzyme bound to a promoter site shows the σ subunit interacting with DNA at the −10 and −35 regions essential to promoter recognition (Figure 29.10). Therefore, the σ subunit is responsible for the specific binding of the RNA polymerase to a promoter site on the template DNA. The σ subunit is generally released when the nascent RNA chain reaches 9 or 10 nucleotides in length. After its release, it can assist initiation by another core enzyme. Thus, the σ subunit acts catalytically.
E. coli has seven distinct σ factors for recognizing several types of promoter sequences in E. coli DNA. The type that recognizes the consensus sequences described earlier is called σ70 because it has a mass of 70 kDa. A different σ factor comes into play when the temperature is raised abruptly. E. coli responds by synthesizing σ32, which recognizes the promoters of heat-shock genes. These promoters exhibit −10 sequences that are somewhat different from the −10 sequence for standard promoters (Figure 29.11). The increased transcription of heat-shock genes leads to the coordinated synthesis of a series of protective proteins. Other σ factors respond to environmental conditions, such as nitrogen starvation. These findings demonstrate that σ plays the key role in determining where RNA polymerase initiates transcription.
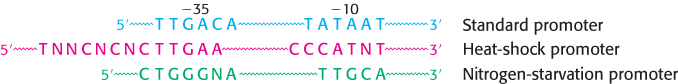
FIGURE 29.11Alternative promoter sequences. A comparison of the consensus sequences of standard, heat-shock, and nitrogen-starvation promoters of E. coli. These promoters are recognized by σ70, σ32, and σ54, respectively.
 Some other bacteria contain a much larger number of σ factors. For example, the genome of the soil bacterium Streptomyces coelicolor encodes more than 60 σ factors recognized on the basis of their amino acid sequences. This repertoire allows these cells to adjust their gene-expression programs to the wide range of conditions, in regard to nutrients and competing organisms, that they may experience.
Some other bacteria contain a much larger number of σ factors. For example, the genome of the soil bacterium Streptomyces coelicolor encodes more than 60 σ factors recognized on the basis of their amino acid sequences. This repertoire allows these cells to adjust their gene-expression programs to the wide range of conditions, in regard to nutrients and competing organisms, that they may experience.
RNA polymerases must unwind the template double helix for transcription to take place
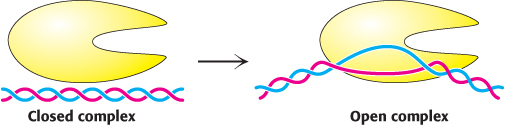
FIGURE 29.12DNA unwinding. The transition from the closed promoter complex to the open promoter complex requires the unwinding of approximately 17 base pairs of DNA.
Although RNA polymerases can search for promoter sites when bound to double-helical DNA, a segment of the DNA double helix must be unwound before synthesis can begin. The transition from the closed promoter complex (in which DNA is double helical) to the open promoter complex (in which a DNA segment is unwound) is an essential event in transcription (Figure 29.12). The free energy necessary to break the bonds between approximately 17 base pairs in the double helix is derived from additional interactions that are possible when the DNA distorts to wrap around the RNA polymerase and from interactions between the single-stranded DNA regions and other parts of the enzyme. These interactions stabilize the open promoter complex and help pull the template strand into the active site. The −35 element remains in a double-helical state, whereas the −10 element is unwound. The stage is now set for the formation of the first phosphodiester bond of the new RNA chain.
Elongation takes place at transcription bubbles that move along the DNA template
The elongation phase of RNA synthesis begins with the formation of the first phosphodiester bond. Repeated cycles of nucleotide addition can take place at this point. However, until about 10 nucleotides have been added, RNA polymerase sometimes releases the short RNA, which dissociates from the DNA. Once RNA polymerase passes this point, the enzyme stays bound to its template until a termination signal is reached. The region containing RNA polymerase, DNA, and nascent RNA corresponds to the transcription bubble (Figure 29.13). The newly synthesized RNA forms a hybrid helix with the template DNA strand. This RNA–DNA helix is about 8 bp long, which corresponds to nearly one turn of a double helix. The 3′-hydroxyl group of the RNA in this hybrid helix is positioned so that it can attack the α-phosphorus atom of an incoming ribonucleoside triphosphate. The core enzyme also contains a binding site for the coding strand of DNA. About 17 bp of DNA are unwound throughout the elongation phase, as in the initiation phase. The transcription bubble moves a distance of 170 Å (17 nm) in a second, which corresponds to a rate of elongation of about 50 nucleotides per second. Although rapid, it is much slower than the rate of DNA synthesis, which is 800 nucleotides per second.
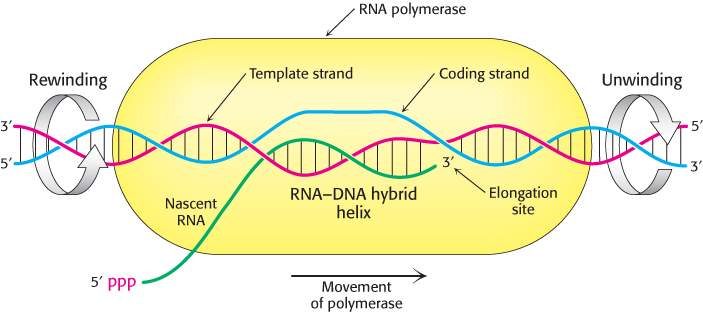
FIGURE 29.13Transcription bubble. A schematic representation of a transcription bubble in the elongation of an RNA transcript. Duplex DNA is unwound at the forward end of RNA polymerase and rewound at its rear end. The RNA–DNA hybrid rotates during elongation.
Sequences within the newly transcribed RNA signal termination
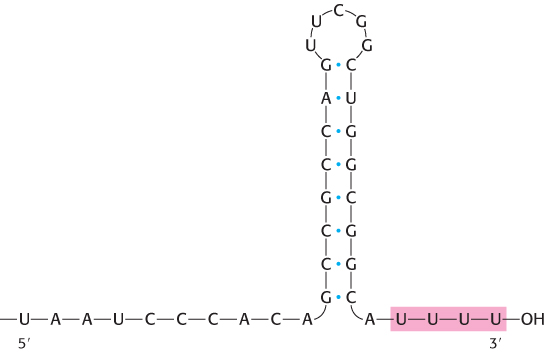
FIGURE 29.14Termination signal. A termination signal found at the 3′ end of an mRNA transcript consists of a series of bases that form a stable stem-loop structure and a series of U residues.
In bacteria, the termination of transcription is as precisely controlled as its initiation. In the termination phase of transcription, the formation of phosphodiester bonds ceases, the RNA–DNA hybrid dissociates, the unwound region of DNA rewinds, and RNA polymerase releases the DNA. What determines where transcription is terminated? The transcribed regions of DNA templates contain stop signals. The simplest one is a palindromic GC-rich region followed by an AT-rich region. The RNA transcript of this DNA palindrome is self-complementary (Figure 29.14). Hence, its bases can pair to form a hairpin structure with a stem and loop, a structure favored by its high content of G and C residues. Guanine–cytosine base pairs are more stable than adenine–thymine pairs because of the extra hydrogen bond in the base pair. This stable hairpin is followed by a sequence of four or more uracil residues, which also are crucial for termination. The RNA transcript ends within or just after them.
How does this combination hairpin–oligo(U) structure terminate transcription? First, RNA polymerase likely pauses immediately after it has synthesized a stretch of RNA that folds into a hairpin. Furthermore, the RNA–DNA hybrid helix produced after the hairpin is unstable because its rU–dA base pairs are the weakest of the four kinds. Hence, the pause in transcription caused by the hairpin permits the weakly bound nascent RNA to dissociate from the DNA template and then from the enzyme. The solitary DNA template strand rejoins its partner to re-form the DNA duplex, and the transcription bubble closes.
Some messenger RNAs directly sense metabolite concentrations
As we shall explore in Chapters 31 and 32, the expression of many genes is controlled in response to the concentrations of metabolites and signaling molecules within cells. One set of control mechanisms depends on the remarkable ability of some mRNA molecules to form special secondary structures, some of which are capable of directly binding small molecules. These structures are termed riboswitches. Consider a riboswitch that controls the synthesis of genes that participate in the biosynthesis of riboflavin in Bacillus subtilis (Figure 29.15). When flavin mononucleotide (FMN), a key intermediate in riboflavin biosynthesis, is present at high concentration, it binds to a conformation of the RNA transcript with an FMN-binding pocket that also includes a hairpin structure that favors premature termination. By trapping the RNA transcript in this termination-favoring conformation, FMN prevents the production of functional mRNA. However, when FMN is present at low concentration, it does not readily bind to RNA, and an alternative conformation forms without the terminator, allowing the production of the full-length mRNA. The occurrence of riboswitches serves as a vivid illustration of how RNAs are capable of forming elaborate, functional structures, though we tend to depict them as simple lines in the absence of specific information.
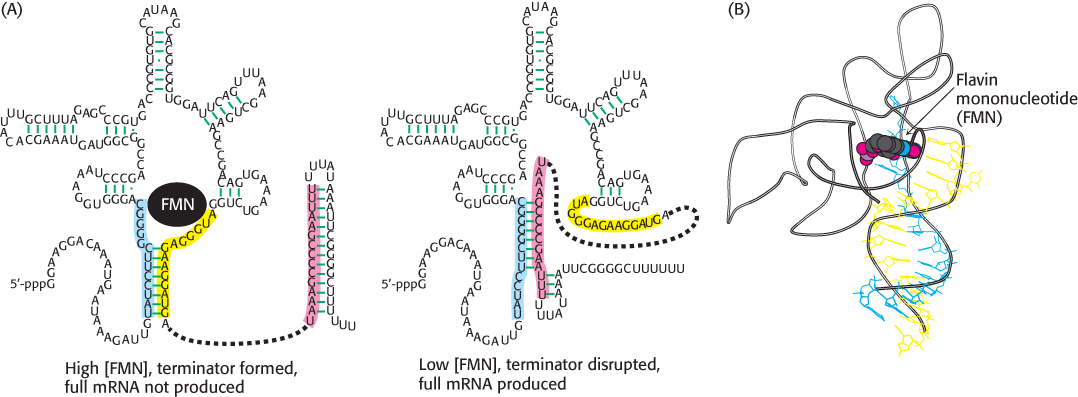
FIGURE 29.15 Riboswitch. (A) The 5′-end of an mRNA that encodes proteins engaged in the production of flavin mononucleotide (FMN) folds to form a structure that is stabilized by binding FMN. This structure includes a terminator that leads to premature termination of the mRNA. At lower concentrations of FMN, an alternative structure that lacks the terminator is formed, leading to the production of full-length mRNA. (B) The three-dimensional structure of a related FMN-binding riboswitch bound to FMN. The blue and yellow stretches correspond to regions highlighted in the same colors in part A. Notice how the yellow strand contacts the bound FMN, stabilizing the structure.
[Drawn from 3F2Q.pdb].
The rho protein helps to terminate the transcription of some genes
RNA polymerase needs no help to terminate transcription at a hairpin followed by several U residues. At other sites, however, termination requires the participation of an additional factor. This discovery was prompted by the observation that some RNA molecules synthesized in vitro by RNA polymerase acting alone are longer than those made in vivo. The missing factor, a protein that caused the correct termination, was isolated and named rho (ρ). Additional information about the action of ρ was obtained by adding this termination factor to an incubation mixture at various times after the initiation of RNA synthesis (Figure 29.16). RNAs with sedimentation coefficients of 10S, 13S, and 17S were obtained when ρ was added at initiation, a few seconds after initiation, and 2 minutes after initiation, respectively. If no ρ was added, transcription yielded a 23S RNA product. It is evident that the template contains at least three termination sites that respond to ρ (yielding 10S, 13S, and 17S RNA) and one termination site that does not (yielding 23S RNA). Thus, specific termination at a site producing 23S RNA can take place in the absence of ρ. However, ρ detects additional termination signals that are not recognized by RNA polymerase alone.
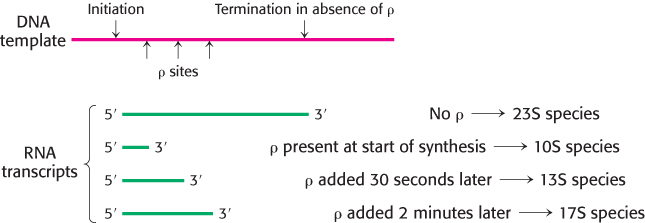
FIGURE 29.16Effect of ρ protein on the size of RNA transcripts.
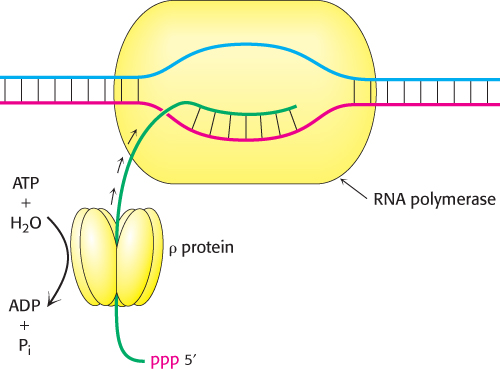
FIGURE 29.17Mechanism for the termination of transcription by ρ protein. This protein is an ATP-dependent helicase that binds the nascent RNA chain and pulls it away from RNA polymerase and the DNA template.
How does ρ provoke the termination of RNA synthesis? A key clue is the finding that ρ hydrolyzes ATP in the presence of single-stranded RNA but not in the presence of DNA or duplex RNA. Hexameric ρ is a helicase, homologous to the helicases that we encountered in our consideration of DNA replication (Section 28.1). A stretch of nucleotides is bound in such a way that the RNA passes through the center of the structure (Figure 29.17). The ρ protein is brought into action by sequences located in the nascent RNA that are rich in cytosine and poor in guanine. The helicase activity of ρ enables the protein to pull the nascent RNA while pursuing RNA polymerase. When ρ catches RNA polymerase at the transcription bubble, it breaks the RNA–DNA hybrid helix by functioning as an RNA–DNA helicase.
Proteins in addition to ρ may provoke termination. For example, the nusA protein enables RNA polymerase in E. coli to recognize a characteristic class of termination sites. A common feature of protein-independent and protein-dependent termination is that the functioning signals lie in newly synthesized RNA rather than in the DNA template.
Some antibiotics inhibit transcription
 Many antibiotics are highly specific inhibitors of biological processes in bacteria. Rifampicin and actinomycin are two antibiotics that inhibit bacterial transcription, although in quite different ways. Rifampicin is a semisynthetic derivative of rifamycins, which are derived from a strain of Amycolatopsis that is related to the bacterium that causes strep throat.
Many antibiotics are highly specific inhibitors of biological processes in bacteria. Rifampicin and actinomycin are two antibiotics that inhibit bacterial transcription, although in quite different ways. Rifampicin is a semisynthetic derivative of rifamycins, which are derived from a strain of Amycolatopsis that is related to the bacterium that causes strep throat.
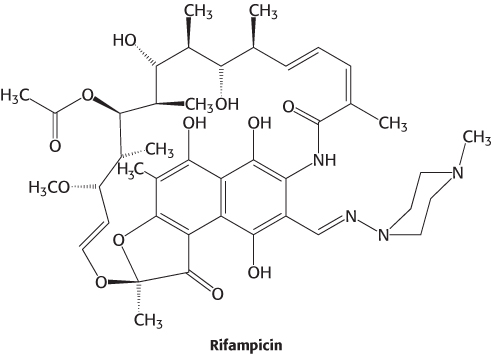
This antibiotic specifically inhibits the initiation of RNA synthesis. Rifampicin interferes with the formation of the first few phosphodiester bonds in the RNA chain. The structure of a complex between a prokaryotic RNA polymerase and rifampicin reveals that the antibiotic blocks the channel into which the RNA–DNA hybrid generated by the enzyme must pass (Figure 29.18). The binding site is 12 Å from the active site itself. Rifampicin can inhibit only the initiation of transcription, not elongation, because the RNA–DNA hybrid present in the enzyme during elongation prevents the antibiotic from binding. The pocket in which rifampicin binds is conserved among bacterial RNA polymerases, but not eukaryotic polymerases, and so rifampicin can be used as an antibiotic in antituberculosis therapy.
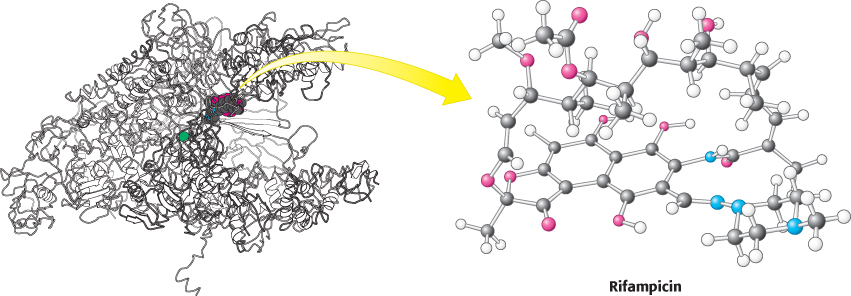
FIGURE 29.18Antibiotic action. Rifampicin binds to a pocket in the channel that is normally occupied by the newly formed RNA–DNA hybrid. Thus, the antibiotic blocks elongation after only two or three nucleotides have been added.
Actinomycin D, a peptide-containing antibiotic from a different strain of Streptomyces, inhibits transcription by an entirely different mechanism. Actinomycin D binds tightly and specifically to double-helical DNA and thereby prevents it from being an effective template for RNA synthesis. The results of spectroscopic, hydrodynamic, and structural studies of complexes of actinomycin D and DNA reveal that the phenoxazone ring of actinomycin slips in between base pairs in DNA (Figure 29.19). This mode of binding is called intercalation. At low concentrations, actinomycin D inhibits transcription without significantly affecting DNA replication or protein synthesis. Hence, actinomycin D is extensively used as a highly specific inhibitor of the formation of new RNA in both prokaryotic and eukaryotic cells. Its ability to inhibit the growth of rapidly dividing cells makes it an effective therapeutic agent in the treatment of some cancers.
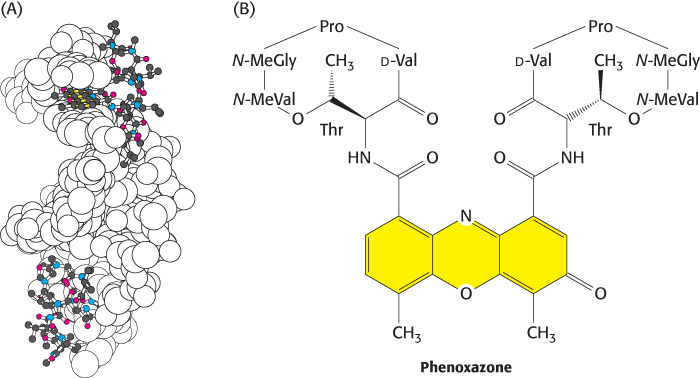
FIGURE 29.19 Actinomycin–DNA complex structure. (A) The structure of a complex between a DNA duplex (shown as a space-filling model) and actinomycin B (shown as a ball-and-stick model). Two actinomycin B molecules are bound in the complex. (B) The structure of actinomycin B showing the phenoxazone ring. Notice how the phenoxazone (yellow) slides between base pairs of the DNA. Abbreviation: Me, methyl.
[Drawn from 1I3W.pdb]
Precursors of transfer and ribosomal RNA are cleaved and chemically modified after transcription in prokaryotes
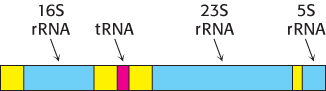
FIGURE 29.20Primary transcript. Cleavage of this transcript produces 5S, 16S, and 23S rRNA molecules and a tRNA molecule. Spacer regions are shown in yellow.
In prokaryotes, messenger RNA molecules undergo little or no modification after synthesis by RNA polymerase. Indeed, many mRNA molecules are translated while they are being transcribed. In contrast, transfer RNA (tRNA) and ribosomal RNA (rRNA) molecules are generated by cleavage and other modifications of nascent RNA chains. For example, in E. coli, the three rRNAs and a tRNA are excised from a single primary RNA transcript that also contains spacer regions (Figure 29.20). Other transcripts contain arrays of several kinds of tRNA or of several copies of the same tRNA. The nucleases that cleave and trim these precursors of rRNA and tRNA are highly precise. Ribonuclease P (RNase P), for example, generates the correct 5′ terminus of all tRNA molecules in E. coli. Sidney Altman and his coworkers showed that this interesting enzyme contains a catalytically active RNA molecule. Ribonuclease III (RNase III) excises 5S, 16S, and 23S rRNA precursors from the primary transcript by cleaving double-helical hairpin regions at specific sites.
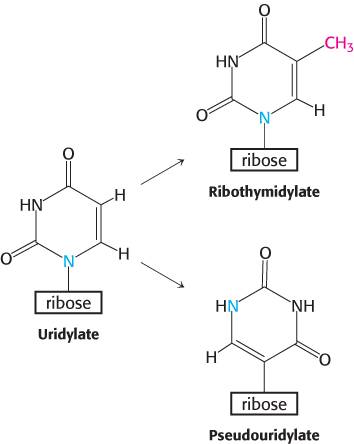
A second type of processing is the addition of nucleotides to the termini of some RNA chains. For example, CCA, a terminal sequence required for the function of all tRNAs, is added to the 3′ ends of tRNA molecules for which this terminal sequence is not encoded in the DNA. The enzyme that catalyzes the addition of CCA is atypical for an RNA polymerase in that it does not use a DNA template. A third type of processing is the modification of bases and ribose units of ribosomal RNAs. In prokaryotes, some bases of rRNA are methylated. Unusual bases are found in all tRNA molecules. They are formed by the enzymatic modification of a standard ribonucleotide in a tRNA precursor. For example, uridylate residues are modified after transcription to form ribothymidylate and pseudouridylate. These modifications generate diversity, allowing greater structural and functional versatility.