9.1
Proteases Facilitate a Fundamentally Difficult Reaction
Peptide bond hydrolysis is an important process in living systems (Chapter 23). Proteins that have served their purpose must be degraded so that their constituent amino acids can be recycled for the synthesis of new proteins. Proteins ingested in the diet must be broken down into small peptides and amino acids for absorption in the gut. Furthermore, as described in detail in Chapter 10, proteolytic reactions are important in regulating the activity of certain enzymes and other proteins.
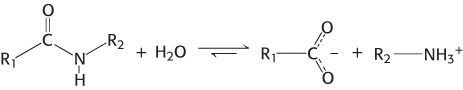
Proteases cleave proteins by a hydrolysis reaction—the addition of a molecule of water to a peptide bond:
Although the hydrolysis of peptide bonds is thermodynamically favorable, such reactions are extremely slow. In the absence of a catalyst, the half-life for the hydrolysis of a typical peptide at neutral pH is estimated to be between 10 and 1000 years. Yet, peptide bonds must be hydrolyzed within milliseconds in some biochemical processes.
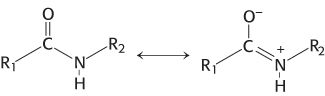
The chemical nature of peptide bonds is responsible for their kinetic stability. Specifically, the resonance structure that accounts for the planarity of peptide bonds (Section 2.2) also makes them resistant to hydrolysis. This resonance structure endows them with partial double-bond character:
The carbon–nitrogen bond is strengthened by its double-bond character. Furthermore, the carbonyl carbon atom is less electrophilic and less susceptible to nucleophilic attack than are the carbonyl carbon atoms in more reactive compounds such as carboxylate esters. Consequently, to promote peptide-bond cleavage, an enzyme must facilitate nucleophilic attack at a normally unreactive carbonyl group.
Chymotrypsin possesses a highly reactive serine residue
A number of proteolytic enzymes participate in the breakdown of proteins in the digestive systems of mammals and other organisms. One such enzyme, chymotrypsin, cleaves peptide bonds selectively on the carboxyl-terminal side of the large hydrophobic amino acids such as tryptophan, tyrosine, phenylalanine, and methionine (Figure 9.1). Chymotrypsin is a good example of the use of covalent catalysis. The enzyme employs a powerful nucleophile to attack the unreactive carbonyl carbon atom of the substrate. This nucleophile becomes covalently attached to the substrate briefly in the course of catalysis.
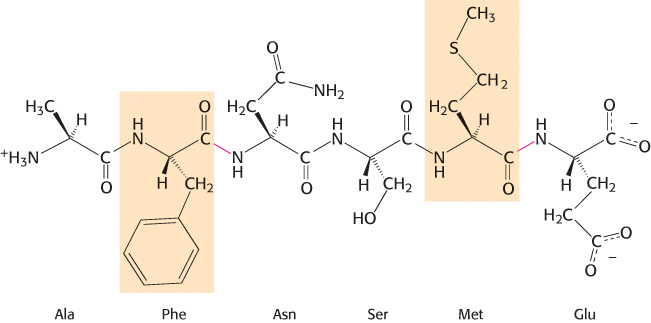
FIGURE 9.1Specificity of chymotrypsin. Chymotrypsin cleaves proteins on the carboxyl side of aromatic or large hydrophobic amino acids (shaded orange). The likely bonds cleaved by chymotrypsin are indicated in red.
What is the nucleophile that chymotrypsin employs to attack the substrate carbonyl carbon atom? A clue came from the fact that chymotrypsin contains an extraordinarily reactive serine residue. Chymotrypsin molecules treated with organofluorophosphates such as diisopropylphosphofluoridate (DIPF) lost all activity irreversibly (Figure 9.2). Only a single residue, serine 195, was modified. This chemical modification reaction suggested that this unusually reactive serine residue plays a central role in the catalytic mechanism of chymotrypsin.
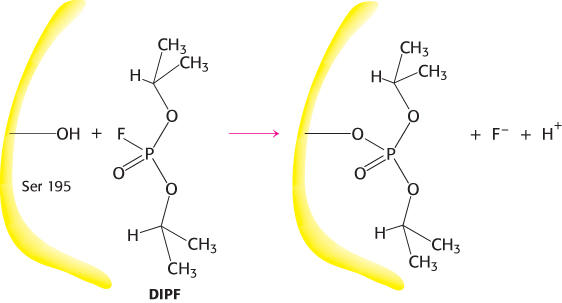
FIGURE 9.2An unusually reactive serine residue in chymotrypsin. Chymotrypsin is inactivated by treatment with diisopropylphosphofluoridate (DIPF), which reacts only with serine 195 among 28 possible serine residues.
Chymotrypsin action proceeds in two steps linked by a covalently bound intermediate
A study of the kinetics of chymotrypsin provided a second clue to its catalytic mechanism. Enzyme kinetics are often easily monitored by having the enzyme act on a substrate analog that forms a colored product. For chymotrypsin, such a chromogenic substrate is N-acetyl-l-phenylalanine p-nitrophenyl ester. This substrate is an ester rather than an amide, but many proteases will also hydrolyze esters. One of the products formed by chymotrypsin’s cleavage of this substrate is p-nitrophenolate, which has a yellow color (Figure 9.3). Measurements of the absorbance of light revealed the amount of p-nitrophenolate being produced.
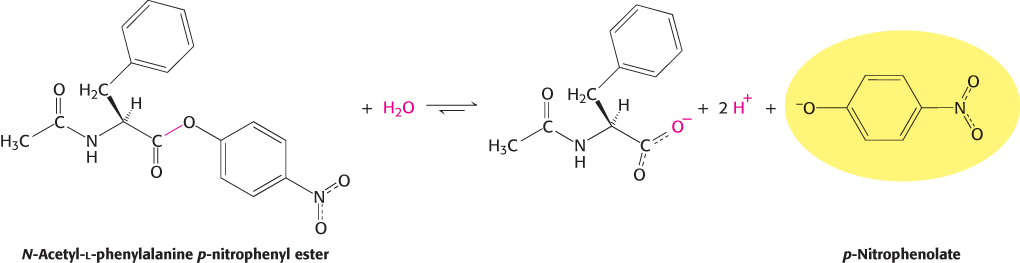
FIGURE 9.3Chromogenic substrate. N-Acetyl-l-phenylalanine p-nitrophenyl ester yields a yellow product, p-nitrophenolate, on cleavage by chymotrypsin. p-Nitrophenolate forms by deprotonation of p-nitrophenol at pH 7.
Under steady-state conditions, the cleavage of this substrate obeys Michaelis–Menten kinetics with a KM of 20 μM and a kcat of 77 s−1. The initial phase of the reaction was examined by using the stopped-flow method, which makes it possible to mix enzyme and substrate and monitor the results within a millisecond. This method revealed an initial rapid burst of colored product, followed by its slower formation as the reaction reached the steady state (Figure 9.4). These results suggest that hydrolysis proceeds in two phases. In the first reaction cycle that takes place immediately after mixing, only the first phase must take place before the colored product is released. In subsequent reaction cycles, both phases must take place. Note that the burst is observed because the first phase is substantially more rapid than the second phase for this substrate.
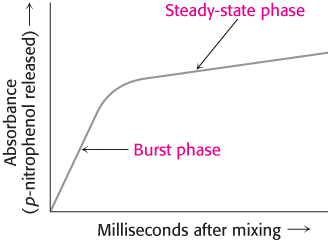
FIGURE 9.4Kinetics of chymotrypsin catalysis. Two phases are evident in the cleaving of N-acetyl-l-phenylalanine p-nitrophenyl ester by chymotrypsin: a rapid burst phase (pre-steady-state) and a steady-state phase.
The two phases are explained by the formation of a covalently bound enzyme–substrate intermediate (Figure 9.5). First, the acyl group of the substrate becomes covalently attached to the enzyme as p-nitrophenolate (or an amine if the substrate is an amide rather than an ester) is released. The enzyme–acyl group complex is called the acyl-enzyme intermediate. Second, the acyl-enzyme intermediate is hydrolyzed to release the carboxylic acid component of the substrate and regenerate the free enzyme. Thus, one molecule of p-nitrophenolate is produced rapidly from each enzyme molecule as the acyl-enzyme intermediate is formed. However, it takes longer for the enzyme to be “reset” by the hydrolysis of the acyl-enzyme intermediate, and both phases are required for enzyme turnover.
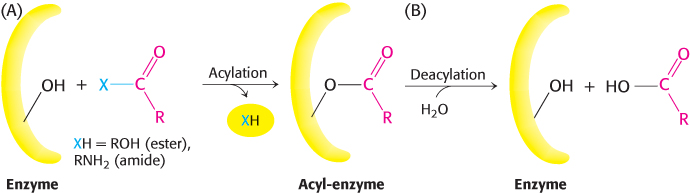
FIGURE 9.5Covalent catalysis. Hydrolysis by chymotrypsin takes place in two phases: (A) acylation to form the acyl-enzyme intermediate followed by (B) deacylation to regenerate the free enzyme.
Serine is part of a catalytic triad that also includes histidine and aspartate
The three-dimensional structure of chymotrypsin revealed that this enzyme is roughly spherical and comprises three polypeptide chains, linked by disulfide bonds. It is synthesized as a single polypeptide, termed chymotrypsinogen, which is activated by the proteolytic cleavage of the polypeptide to yield the three chains (Section 10.4). The active site of chymotrypsin, marked by serine 195, lies in a cleft on the surface of the enzyme (Figure 9.6). The structure of the active site explained the special reactivity of serine 195 (Figure 9.7). The side chain of serine 195 is hydrogen bonded to the imidazole ring of histidine 57. The —NH group of this imidazole ring is, in turn, hydrogen bonded to the carboxylate group of aspartate 102. This constellation of residues is referred to as the catalytic triad. How does this arrangement of residues lead to the high reactivity of serine 195? The histidine residue serves to position the serine side chain and to polarize its hydroxyl group so that it is poised for deprotonation. In the presence of the substrate, the histidine residue accepts the proton from the serine 195 hydroxyl group. In doing so, the histidine acts as a general base catalyst. The withdrawal of the proton from the hydroxyl group generates an alkoxide ion, which is a much more powerful nucleophile than is an alcohol. The aspartate residue helps orient the histidine residue and make it a better proton acceptor through hydrogen bonding and electrostatic effects.
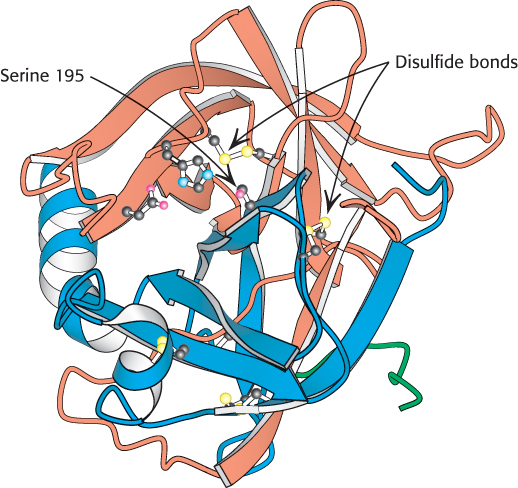
 FIGURE 9.6 Location of the active site in chymotrypsin. Chymotrypsin consists of three chains, shown in ribbon form in orange, blue, and green. The side chains of the catalytic triad residues are shown as ball-and-stick representations. Notice these side chains, including serine 195, lining the active site in the upper half of the structure. Also notice two intrastrand and two interstrand disulfide bonds in various locations throughout the molecule.
FIGURE 9.6 Location of the active site in chymotrypsin. Chymotrypsin consists of three chains, shown in ribbon form in orange, blue, and green. The side chains of the catalytic triad residues are shown as ball-and-stick representations. Notice these side chains, including serine 195, lining the active site in the upper half of the structure. Also notice two intrastrand and two interstrand disulfide bonds in various locations throughout the molecule.
[Drawn from 1GCT.pdb.]
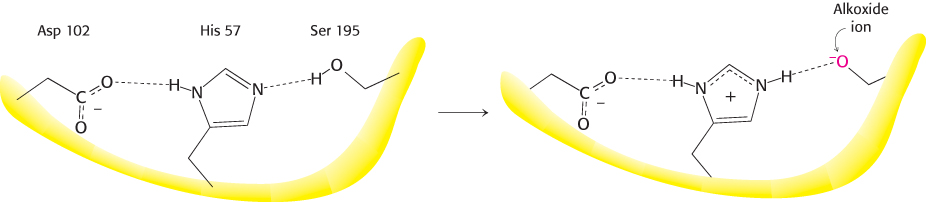
FIGURE 9.7The catalytic triad. The catalytic triad, shown on the left, converts serine 195 into a potent nucleophile, as illustrated on the right.
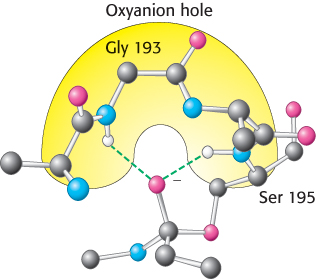
FIGURE 9.9The oxyanion hole. The structure stabilizes the tetrahedral intermediate of the chymotrypsin reaction. Notice that hydrogen bonds (shown in green) link peptide NH groups and the negatively charged oxygen atom of the intermediate.
These observations suggest a mechanism for peptide hydrolysis (Figure 9.8). After substrate binding (step 1), the reaction begins with the oxygen atom of the side chain of serine 195 making a nucleophilic attack on the carbonyl carbon atom of the target peptide bond (step 2). There are now four atoms bonded to the carbonyl carbon, arranged as a tetrahedron, instead of three atoms in a planar arrangement. This inherently unstable tetrahedral intermediate bears a formal negative charge on the oxygen atom derived from the carbonyl group. This charge is stabilized by interactions with NH groups from the protein in a site termed the oxyanion hole (Figure 9.9). These interactions also help stabilize the transition state that precedes the formation of the tetrahedral intermediate. This tetrahedral intermediate collapses to generate the acyl-enzyme (step 3). This step is facilitated by the transfer of the proton being held by the positively charged histidine residue to the amino group formed by cleavage of the peptide bond. The amine component is now free to depart from the enzyme (step 4), completing the first stage of the hydrolytic reaction—acylation of the enzyme. Such acyl-enzyme intermediates have even been observed using X-ray crystallography by trapping them through adjustment of conditions such as the nature of the substrate, pH, or temperature.
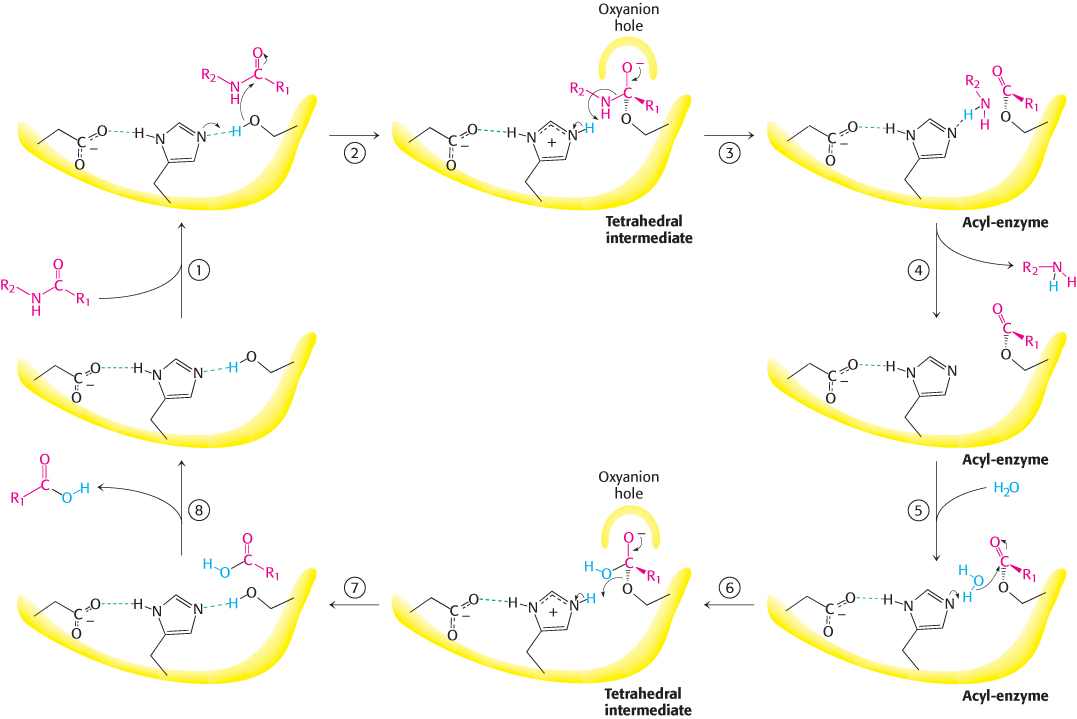
FIGURE 9.8Peptide hydrolysis by chymotrypsin. The mechanism of peptide hydrolysis illustrates the principles of covalent and acid–base catalysis. The reaction proceeds in eight steps: (1) substrate binding, (2) nucleophilic attack of serine on the peptide carbonyl group, (3) collapse of the tetrahedral intermediate, (4) release of the amine component, (5) water binding, (6) nucleophilic attack of water on the acyl-enzyme intermediate, (7) collapse of the tetrahedral intermediate; and (8) release of the carboxylic acid component. The dashed green lines represent hydrogen bonds.
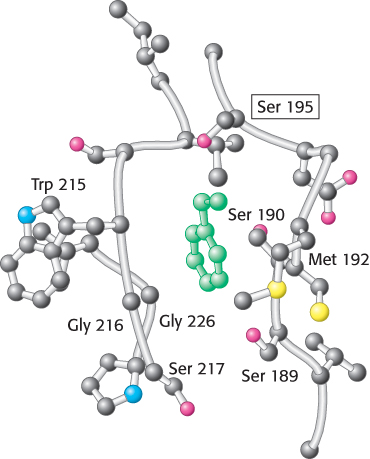
FIGURE 9.10Specificity pocket of chymotrypsin. Notice that this pocket is lined with hydrophobic residues and is deep, favoring the binding of residues with long hydrophobic side chains such as phenylalanine (shown in green). The active-site serine residue (serine 195) is positioned to cleave the peptide backbone between the residue bound in the pocket and the next residue in the sequence. The key amino acids that constitute the binding site are identified.
The next stage—deacylation—begins when a water molecule takes the place occupied earlier by the amine component of the substrate (step 5). The ester group of the acyl-enzyme is now hydrolyzed by a process that essentially repeats steps 2 through 4. Now acting as a general acid catalyst, histidine 57 draws a proton away from the water molecule. The resulting OH− ion attacks the carbonyl carbon atom of the acyl group, forming a tetrahedral intermediate (step 6). This structure breaks down to form the carboxylic acid product (step 7). Finally, the release of the carboxylic acid product (step 8) readies the enzyme for another round of catalysis.
This mechanism accounts for all characteristics of chymotrypsin action except the observed preference for cleaving the peptide bonds just past residues with large, hydrophobic side chains. Examination of the three-dimensional structure of chymotrypsin with substrate analogs and enzyme inhibitors revealed the presence of a deep hydrophobic pocket, called the S1 pocket, into which the long, uncharged side chains of residues such as phenylalanine and tryptophan can fit. The binding of an appropriate side chain into this pocket positions the adjacent peptide bond into the active site for cleavage (Figure 9.10). The specificity of chymotrypsin depends almost entirely on which amino acid is directly on the amino-terminal side of the peptide bond to be cleaved. Other proteases have more-complex specificity patterns. Such enzymes have additional pockets on their surfaces for the recognition of other residues in the substrate. Residues on the amino-terminal side of the scissile bond (the bond to be cleaved) are labeled P1, P2, P3, and so forth, heading away from the scissile bond (Figure 9.11). Likewise, residues on the carboxyl side of the scissile bond are labeled 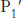 ,
, 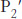 ,
, 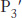 , and so forth. The corresponding sites on the enzyme are referred to as S1, S2 or
, and so forth. The corresponding sites on the enzyme are referred to as S1, S2 or 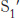 ,
,  , and so forth.
, and so forth.
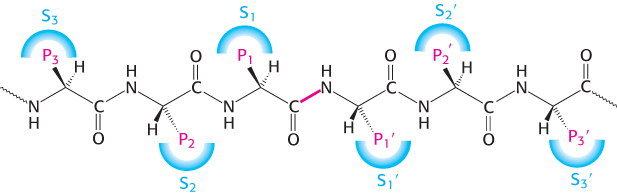
FIGURE 9.11Specificity nomenclature for protease–substrate interactions. The potential sites of interaction of the substrate with the enzyme are designated P (shown in red), and corresponding binding sites on the enzyme are designated S. The scissile bond (also shown in red) is the reference point.
Catalytic triads are found in other hydrolytic enzymes
 Many other peptide-cleaving proteins have subsequently been found to contain catalytic triads similar to that discovered in chymotrypsin. Some, such as trypsin and elastase, are obvious homologs of chymotrypsin. The sequences of these proteins are approximately 40% identical with that of chymotrypsin, and their overall structures are quite similar (Figure 9.12). These proteins operate by mechanisms identical with that of chymotrypsin. However, the three enzymes differ markedly in substrate specificity. Chymotrypsin cleaves at the peptide bond after residues with an aromatic or long nonpolar side chain. Trypsin cleaves at the peptide bond after residues with long, positively charged side chains—namely, arginine and lysine. Elastase cleaves at the peptide bond after amino acids with small side chains—such as alanine and serine. Comparison of the S1 pockets of these enzymes reveals that these different specificities are due to small structural differences. In trypsin, an aspartate residue (Asp 189) is present at the bottom of the S1 pocket in place of a serine residue in chymotrypsin. The aspartate residue attracts and stabilizes a positively charged arginine or lysine residue in the substrate. In elastase, two residues at the top of the pocket in chymotrypsin and trypsin are replaced by much bulkier valine residues (Val 190 and Val 216). These residues close off the mouth of the pocket so that only small side chains can enter (Figure 9.13).
Many other peptide-cleaving proteins have subsequently been found to contain catalytic triads similar to that discovered in chymotrypsin. Some, such as trypsin and elastase, are obvious homologs of chymotrypsin. The sequences of these proteins are approximately 40% identical with that of chymotrypsin, and their overall structures are quite similar (Figure 9.12). These proteins operate by mechanisms identical with that of chymotrypsin. However, the three enzymes differ markedly in substrate specificity. Chymotrypsin cleaves at the peptide bond after residues with an aromatic or long nonpolar side chain. Trypsin cleaves at the peptide bond after residues with long, positively charged side chains—namely, arginine and lysine. Elastase cleaves at the peptide bond after amino acids with small side chains—such as alanine and serine. Comparison of the S1 pockets of these enzymes reveals that these different specificities are due to small structural differences. In trypsin, an aspartate residue (Asp 189) is present at the bottom of the S1 pocket in place of a serine residue in chymotrypsin. The aspartate residue attracts and stabilizes a positively charged arginine or lysine residue in the substrate. In elastase, two residues at the top of the pocket in chymotrypsin and trypsin are replaced by much bulkier valine residues (Val 190 and Val 216). These residues close off the mouth of the pocket so that only small side chains can enter (Figure 9.13).
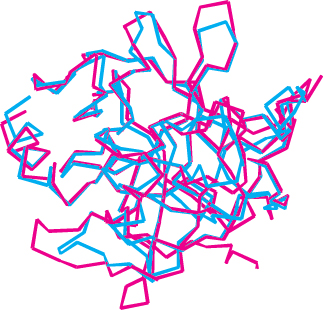
FIGURE 9.12 Structural similarity of trypsin and chymotrypsin. An overlay of the structure of chymotrypsin (red) on that of trypsin (blue) is shown. Notice the high degree of similarity. Only α-carbon-atom positions are shown. The mean deviation in position between corresponding α-carbon atoms is 1.7 Å.
[Drawn from 5PTP.pdb and 1GCT.pdb.]
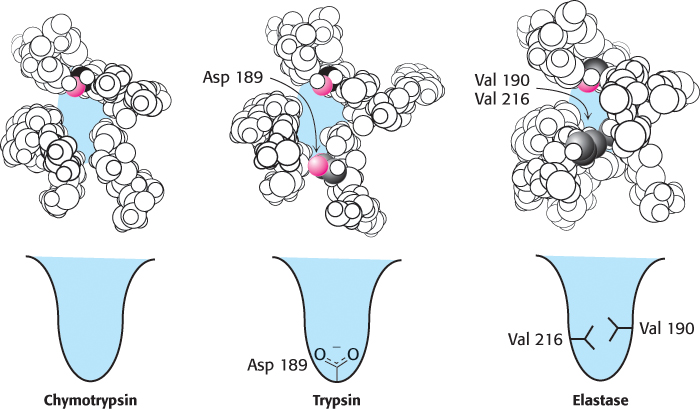
FIGURE 9.13The S1 pockets of chymotrypsin, trypsin, and elastase. Certain residues play key roles in determining the specificity of these enzymes. The side chains of these residues, as well as those of the active-site serine residues, are shown in color.
Other members of the chymotrypsin family include a collection of proteins that take part in blood clotting, to be discussed in Chapter 10, as well as the tumor marker protein prostate-specific antigen (PSA). In addition, a wide range of proteases found in bacteria, viruses, and plants belong to this clan.
Other enzymes that are not homologs of chymotrypsin have been found to contain very similar active sites. As noted in Chapter 6, the presence of very similar active sites in these different protein families is a consequence of convergent evolution. Subtilisin, a protease in bacteria such as Bacillus amyloliquefaciens, is a particularly well characterized example. The active site of this enzyme includes both the catalytic triad and the oxyanion hole. However, one of the NH groups that forms the oxyanion hole comes from the side chain of an asparagine residue rather than from the peptide backbone (Figure 9.14). Subtilisin is the founding member of another large family of proteases that includes representatives from Archaea, Bacteria, and Eukarya.
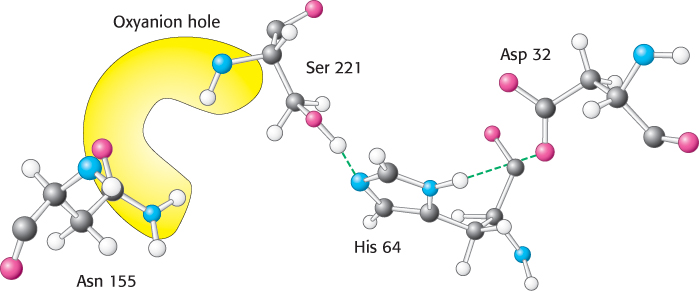
FIGURE 9.14The catalytic triad and oxyanion hole of subtilisin. Notice the two enzyme NH groups (both in the backbone and in the side chain of Asn 155) located in the oxyanion hole. The NH groups will stabilize a negative charge that develops on the peptide bond attacked by nucleophilic serine 221 of the catalytic triad.
Finally, other proteases have been discovered that contain an active-site serine or threonine residue that is activated not by a histidine–aspartate pair but by a primary amino group from the side chain of lysine or by the N-terminal amino group of the polypeptide chain.
Thus, the catalytic triad in proteases has emerged at least three times in the course of evolution. We can conclude that this catalytic strategy must be an especially effective approach to the hydrolysis of peptides and related bonds.
The catalytic triad has been dissected by site-directed mutagenesis
How can we test the validity of the mechanism proposed for the catalytic triad? One way is to test the contribution of individual amino acid residues to the catalytic power of a protease by using site-directed mutagenesis (Section 5.2). Subtilisin has been extensively studied by this method. Each of the residues within the catalytic triad, consisting of aspartic acid 32, histidine 64, and serine 221, has been individually converted into alanine, and the ability of each mutant enzyme to cleave a model substrate has been examined (Figure 9.15).
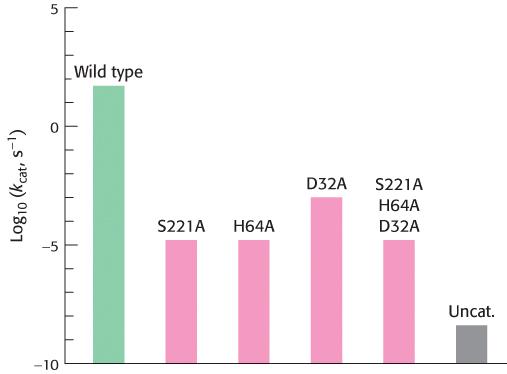
FIGURE 9.15Site-directed mutagenesis of subtilisin. Residues of the catalytic triad were mutated to alanine, and the activity of the mutated enzyme was measured. Mutations in any component of the catalytic triad cause a dramatic loss of enzyme activity. Note that the activity is displayed on a logarithmic scale. The mutations are identified as follows: the first letter is the one-letter abbreviation for the amino acid being altered; the number identifies the position of the residue in the primary structure; and the second letter is the one-letter abbreviation for the amino acid replacing the original one. Uncat. refers to the estimated rate for the uncatalyzed reaction.
As expected, the conversion of active-site serine 221 into alanine dramatically reduced catalytic power; the value of kcat fell to less than one millionth of its value for the wild-type enzyme. The value of KM was essentially unchanged; its increase by no more than a factor of two indicated that substrate continued to bind normally. The mutation of histidine 64 to alanine reduced catalytic power to a similar degree. The conversion of aspartate 32 into alanine reduced catalytic power by less, although the value of kcat still fell to less than 0.005% of its wild-type value. The simultaneous conversion of all three residues into alanine was no more deleterious than the conversion of serine or histidine alone. These observations support the notion that the catalytic triad and, particularly, the serine–histidine pair act together to generate a nucleophile of sufficient power to attack the carbonyl carbon atom of a peptide bond. Despite the reduction in their catalytic power, the mutated enzymes still hydrolyze peptides a thousand times as fast as buffer at pH 8.6.
Site-directed mutagenesis also offered a way to probe the importance of the oxyanion hole for catalysis. The mutation of asparagine 155 to glycine eliminated the side-chain NH group from the oxyanion hole of subtilisin. The elimination of the NH group reduced the value of kcat to 0.2% of its wild-type value but increased the value of KM by only a factor of two. These observations demonstrate that the NH group of the asparagine residue plays a significant role in stabilizing the tetrahedral intermediate and the transition state leading to it.
Cysteine, aspartyl, and metalloproteases are other major classes of peptide-cleaving enzymes
Not all proteases utilize strategies based on activated serine residues. Classes of proteins have been discovered that employ three alternative approaches to peptide-bond hydrolysis (Figure 9.16). These classes are the (1) cysteine proteases, (2) aspartyl proteases, and (3) metalloproteases. In each case, the strategy is to generate a nucleophile that attacks the peptide carbonyl group (Figure 9.17).
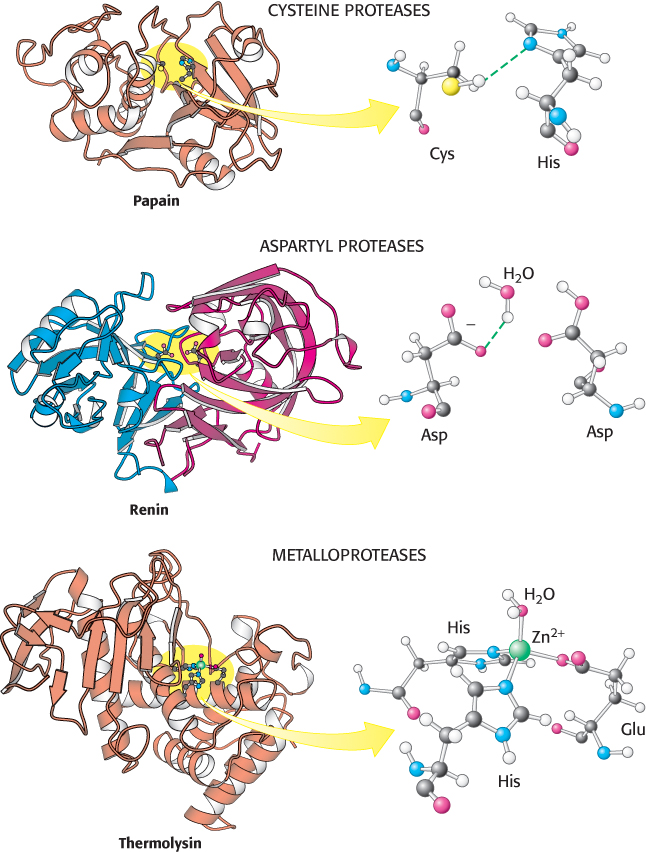
FIGURE 9.16 Three classes of proteases and their active sites. These examples of a cysteine protease, an aspartyl protease, and a metalloprotease use a histidine-activated cysteine residue, an aspartate-activated water molecule, and a metal-activated water molecule, respectively, as the nucleophile. The two halves of renin are in blue and red to highlight the approximate twofold symmetry of aspartyl proteases. Notice how different these active sites are despite the similarity in the reactions they catalyze.
[Drawn from 1PPN.pdb.; 1HRN.pdb; 1LND.pdb.]
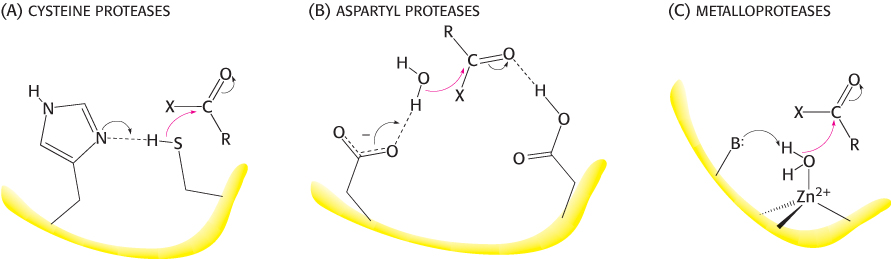
FIGURE 9.17The activation strategies for three classes of proteases. The peptide carbonyl group is attacked by (A) a histidine-activated cysteine in the cysteine proteases, (B) an aspartate-activated water molecule in the aspartyl proteases, and (C) a metal-activated water molecule in the metalloproteases. For the metalloproteases, the letter B represents a base (often glutamate) that helps deprotonate the metal-bound water.
The strategy used by the cysteine proteases is most similar to that used by the chymotrypsin family. In these enzymes, a cysteine residue, activated by a histidine residue, plays the role of the nucleophile that attacks the peptide bond (Figure 9.17) in a manner quite analogous to that of the serine residue in serine proteases. Because the sulfur atom in cysteine is inherently a better nucleophile than is the oxygen atom in serine, cysteine proteases appear to require only this histidine residue in addition to cysteine and not the full catalytic triad. A well-studied example of these proteins is papain, an enzyme purified from the fruit of the papaya. Mammalian proteases homologous to papain have been discovered, most notably the cathepsins, proteins having a role in the immune system and other systems. The cysteine-based active site arose independently at least twice in the course of evolution; the caspases, enzymes that play a major role in apoptosis (a genetically programmed cell death pathway), have active sites similar to that of papain, but their overall structures are unrelated.
The second class comprises the aspartyl proteases. The central feature of the active sites is a pair of aspartic acid residues that act together to allow a water molecule to attack the peptide bond. One aspartic acid residue (in its deprotonated form) activates the attacking water molecule by poising it for deprotonation. The other aspartic acid residue (in its protonated form) polarizes the peptide carbonyl group so that it is more susceptible to attack (Figure 9.17). Members of this class include renin, an enzyme involved in the regulation of blood pressure, and the digestive enzyme pepsin. These proteins possess approximate twofold symmetry. A likely scenario is that two copies of a gene for the ancestral enzyme fused to form a single gene that encoded a single-chain enzyme. Each copy of the gene would have contributed an aspartate residue to the active site. The individual chains are now joined to make a single chain in most aspartyl proteases, whereas the proteases present in human immunodeficiency virus (HIV) and other retroviruses comprise dimers of identical chains (Figure 9.18). This observation is consistent with the idea that larger aspartyl proteases may have evolved by fusion of separate subunits.
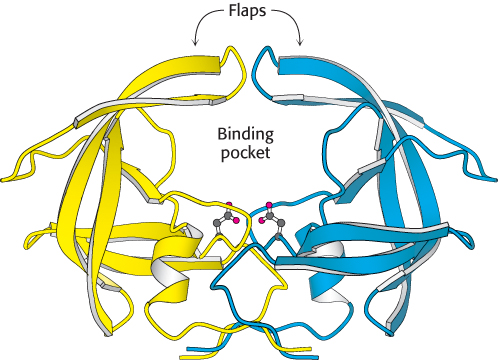
FIGURE 9.18 HIV protease, a dimeric aspartyl protease. The protease is a dimer of identical subunits, shown in blue and yellow, consisting of 99 amino acids each. Notice the placement of active-site aspartic acid residues, one from each chain, which are shown as ball-and-stick structures. The flaps will close down on the binding pocket after substrate has been bound.
[Drawn from 3PHV.pdb.]
The metalloproteases constitute the final major class of peptide-cleaving enzymes. The active site of such a protein contains a bound metal ion, almost always zinc, that activates a water molecule to act as a nucleophile to attack the peptide carbonyl group. The bacterial enzyme thermolysin and the digestive enzyme carboxypeptidase A are classic examples of the zinc proteases. Thermolysin, but not carboxypeptidase A, is a member of a large and diverse family of homologous zinc proteases that includes the matrix metalloproteases, enzymes that catalyze the reactions in tissue remodeling and degradation.
In each of these three classes of enzymes, the active site includes features that act to (1) activate a water molecule or another nucleophile, (2) polarize the peptide carbonyl group, and (3) stabilize a tetrahedral intermediate (Figure 9.17).
Protease inhibitors are important drugs
 Several important drugs are protease inhibitors. For example, captopril, used to regulate blood pressure, is one of many inhibitors of the angiotensin-converting enzyme (ACE), a metalloprotease. Indinavir (Crixivan), retrovir, and more than 20 other compounds used in the treatment of AIDS are inhibitors of HIV protease (Figure 9.18), an aspartyl protease. HIV protease cleaves multidomain viral proteins into their active forms; blocking this process completely prevents the virus from being infectious. HIV protease inhibitors, in combination with inhibitors of other key HIV enzymes, dramatically reduced deaths due to AIDS, assuming that the cost of the treatment could be covered (Figure 36.21). In many cases, these drugs have converted AIDS from a death sentence to a treatable chronic disease.
Several important drugs are protease inhibitors. For example, captopril, used to regulate blood pressure, is one of many inhibitors of the angiotensin-converting enzyme (ACE), a metalloprotease. Indinavir (Crixivan), retrovir, and more than 20 other compounds used in the treatment of AIDS are inhibitors of HIV protease (Figure 9.18), an aspartyl protease. HIV protease cleaves multidomain viral proteins into their active forms; blocking this process completely prevents the virus from being infectious. HIV protease inhibitors, in combination with inhibitors of other key HIV enzymes, dramatically reduced deaths due to AIDS, assuming that the cost of the treatment could be covered (Figure 36.21). In many cases, these drugs have converted AIDS from a death sentence to a treatable chronic disease.
Indinavir resembles the peptide substrate of the HIV protease. Indinavir is constructed around an alcohol that mimics the tetrahedral intermediate; other groups are present to bind into the S2, S1,
, and
recognition sites on the enzyme (Figure 9.19). X-ray crystallographic studies revealed that, in the active site, indinavir adopts a confirmation that approximates the twofold symmetry of the enzyme (Figure 9.20).
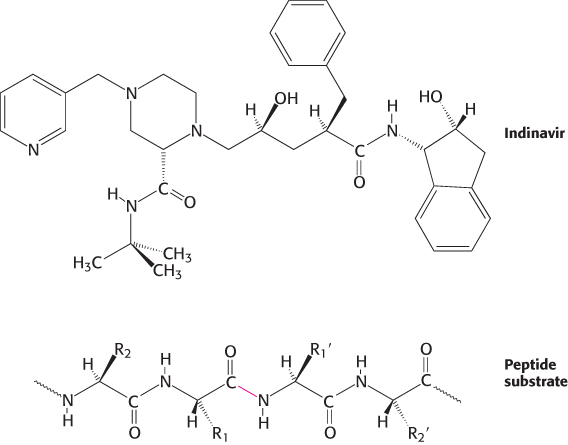
FIGURE 9.19Indinavir, an HIV protease inhibitor. The structure of indinavir (Crixivan) is shown in comparison with that of a peptide substrate of HIV protease. The scissile bond in the substrate is highlighted in red.
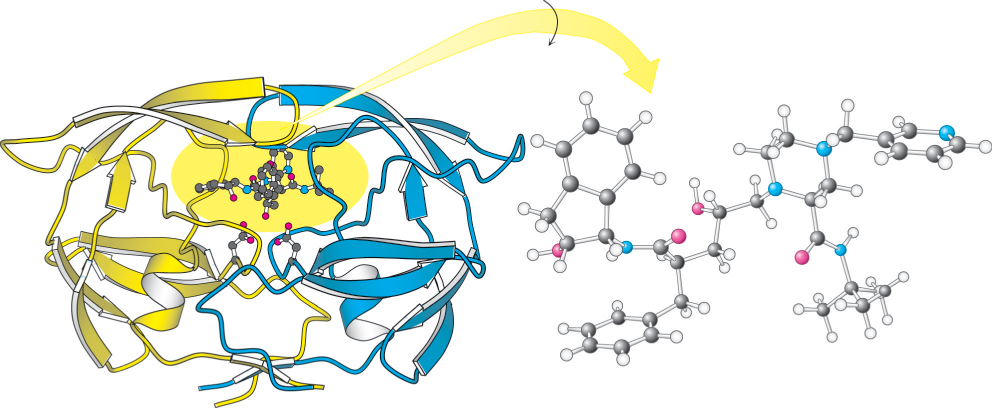
FIGURE 9.20 HIV protease–indinavir complex. (Left) The HIV protease is shown with the inhibitor indinavir bound at the active site. Notice the twofold symmetry of the enzyme structure. (Right) The drug has been rotated to reveal its approximately twofold symmetric conformation.
[Drawn from 1HSH.pdb.]
The active site of HIV protease is covered by two flexible flaps that fold down on top of the bound inhibitor. The OH group of the central alcohol interacts with the two aspartate residues of the active site. In addition, two carbonyl groups of the inhibitor are hydrogen bonded to a water molecule (not shown in Figure 9.20), which, in turn, is hydrogen bonded to a peptide NH group in each of the flaps. This interaction of the inhibitor with water and the enzyme is not possible within cellular aspartyl proteases such as renin. Thus, the interaction may contribute to the specificity of indinavir for HIV protease. To prevent side effects, protease inhibitors used as drugs must be specific for one enzyme without inhibiting other proteins within the body.