Restriction enzymes and DNA ligase are key tools in forming recombinant DNA molecules
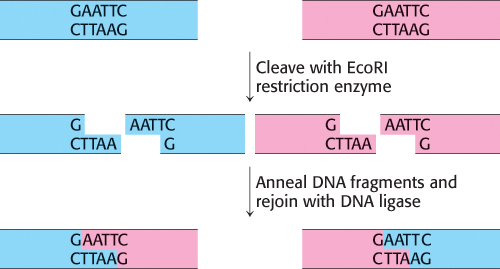
FIGURE 5.9Joining of DNA molecules by the cohesive-end method. Two DNA molecules, cleaved with a common restriction enzyme such as EcoRI, can be ligated to form recombinant molecules.
Let us begin by seeing how novel DNA molecules can be constructed in the laboratory. An essential tool for the manipulation of recombinant DNA is a vector, a DNA molecule that can replicate autonomously in an appropriate host organism. Vectors are designed to enable the rapid, covalent insertion of DNA fragments of interest. Plasmids (naturally occurring circles of DNA that act as accessory chromosomes in bacteria) and bacteriophage lambda (λ phage), a virus, are choice vectors for cloning in E. coli. The vector can be prepared for accepting a new DNA fragment by cleaving it at a single specific site with a restriction enzyme. For example, the plasmid pSC101, a 9.9-kb double-helical circular DNA molecule, is split at a unique site by the EcoRI restriction enzyme. The staggered cuts made by this enzyme produce complementary single-stranded ends, which have specific affinity for each other and hence are known as cohesive or sticky ends. Any DNA fragment can be inserted into this plasmid if it has the same cohesive ends. Such a fragment can be extracted from a larger piece of DNA by using the same restriction enzyme as was used to open the plasmid DNA (Figure 5.9).
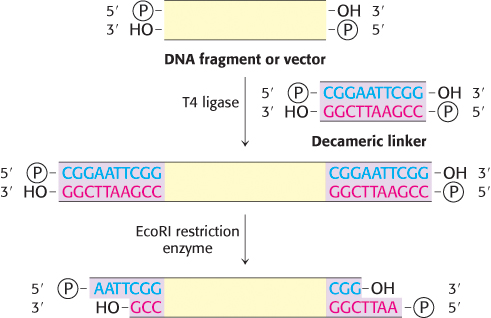
FIGURE 5.10Formation of cohesive ends. Cohesive ends can be formed by the addition and cleavage of a chemically synthesized linker.
The single-stranded ends of the fragment are then complementary to those of the cut plasmid. The DNA fragment and the cut plasmid can be annealed and then joined by DNA ligase, which catalyzes the formation of a phosphodiester bond at a break in a DNA chain. DNA ligase requires a free 3′-hydroxyl group and a 5′-phosphoryl group. Furthermore, the chains joined by ligase must be in a double helix. An energy source such as ATP or NAD+ is required for the joining reaction, as will be discussed in Chapter 28.
What if the target DNA is not naturally flanked by the appropriate restriction sites? How is the fragment cut and annealed to the vector? The cohesive-end method for joining DNA molecules can still be used in these cases by adding a short, chemically synthesized DNA linker that can be cleaved by restriction enzymes. First, the linker is covalently joined to the ends of a DNA fragment. For example, the 5′ ends of a decameric linker and a DNA molecule are phosphorylated by polynucleotide kinase and then joined by the ligase from T4 phage (Figure 5.10). This ligase can form a covalent bond between blunt-ended double-helical DNA molecules. Cohesive ends are produced when these terminal extensions are cut by an appropriate restriction enzyme. Thus, cohesive ends corresponding to a particular restriction enzyme can be added to virtually any DNA molecule. We see here the fruits of combining enzymatic and synthetic chemical approaches in crafting new DNA molecules.
Plasmids and λ phage are choice vectors for DNA cloning in bacteria
Many plasmids and bacteriophages have been ingeniously modified by researchers to enhance the delivery of recombinant DNA molecules into bacteria and to facilitate the genetic selection of bacteria harboring these vectors. As already mentioned, plasmids are circular double-stranded DNA molecules that occur naturally in some bacteria. They range in size from two to several hundred kilobases. Plasmids carry genes for the inactivation of antibiotics, the production of toxins, and the breakdown of natural products. These accessory chromosomes can replicate independently of the host chromosome. In contrast with the host genome, they are dispensable under certain conditions. A bacterial cell may have no plasmids at all or it may house as many as 20 copies of a naturally-occurring plasmid.
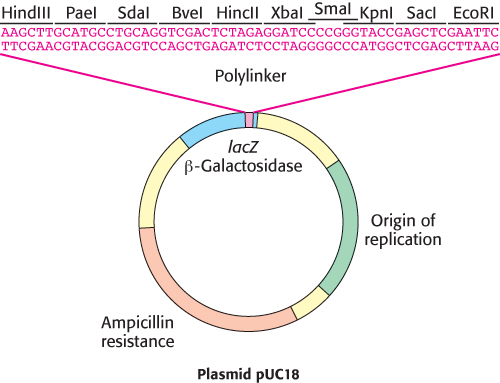
FIGURE 5.11A polylinker in the plasmid pUC18. The plasmid pUC18 includes a polylinker within an essential fragment of the β-galactosidase gene (often called the lacZ gene). Insertion of a DNA fragment into one of the many restriction sites within this polylinker can be detected by the absence of β-galactosidase activity.
Many plasmids have been optimized for a particular experimental task. Some engineered plasmids, for example, can achieve nearly a thousand copies per bacterial cell. One class of plasmids, known as cloning vectors, is particularly suitable for the facile insertion and replication of a collection of DNA fragments. These vectors often feature a polylinker region that includes many unique restriction sites within its sequence. This polylinker can be cleaved with many different restriction enzymes or combinations of enzymes, providing great versatility in the DNA fragments that can be inserted. In addition, these plasmids contain reporter genes, which encode rapidly-detectable markers such as antibiotic-resistance enzymes or fluorescent proteins. Creative placement of these reporter genes within these plasmids enables the rapid identification of those vectors that harbor the desired DNA insert. For example, in the cloning vector pUC18, insertion of DNA in the polylinker region (Figure 5.11) disrupts the lacZα gene, an effect called insertional inactivation. This gene encodes an essential fragment of the protein β-galactosidase, an enzyme which naturally cleaves the milk sugar galactose (Section 11.2). β-Galactosidase also cleaves the synthetic substrate X-gal, releasing a blue dye. Bacterial cells containing a DNA insert at the polylinker will no longer produce the dye in the presence of X-gal, and are readily identified by their white color (Figure 5.12).

FIGURE 5.12Insertional inactivation. Successful insertion of DNA fragments into the polylinker region of pUC18 will result in the disruption of the β-galactosidase gene. Bacterial colonies that harbor such plasmids will no longer convert X-gal into a colored product, and will appear white on the plate.
Another class of plasmids has been optimized for use as expression vectors for the production of large amounts of protein. In addition to antibiotic-resistance genes, they contain promoter sequences designed to drive the transcription of large amounts of a protein-coding DNA sequence. In addition to polylinkers, these vectors often contain sequences flanking the cloning site that simplify the addition of fusion tags to the protein of interest (Section 3.1), greatly facilitating the purification of the overexpressed protein.
Another widely used vector, λ phage, enjoys a choice of life styles: this bacteriophage can destroy its host or it can become part of its host (Figure 5.13). In the lytic pathway, viral functions are fully expressed: viral DNA and proteins are quickly produced and packaged into virus particles, leading to the lysis (destruction) of the host cell and the sudden appearance of about 100 progeny virus particles, or virions. In the lysogenic pathway, the phage DNA becomes inserted into the host-cell genome and can be replicated together with host-cell DNA for many generations, remaining inactive. Certain environmental changes can trigger the expression of this dormant viral DNA, which leads to the formation of progeny viruses and lysis of the host. Large segments of the 48-kb DNA of λ phage are not essential for productive infection and can be replaced by foreign DNA, thus making λ phage an ideal vector.
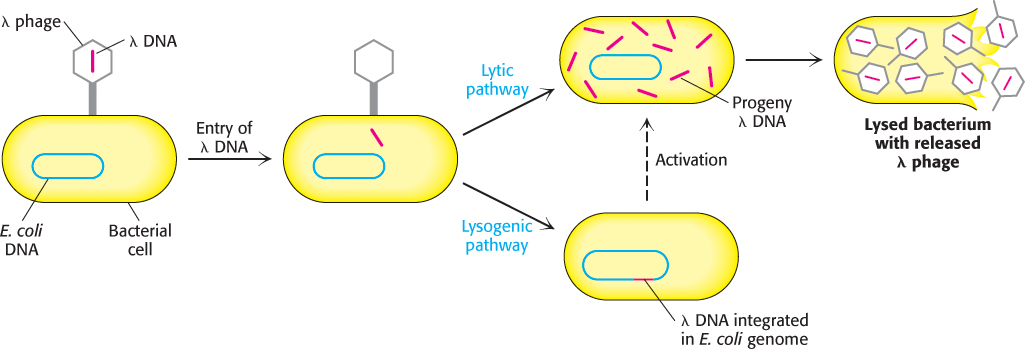
FIGURE 5.13Alternative infection modes for λ phage. Lambda phage can multiply within a host and lyse it (lytic pathway) or its DNA can become integrated into the host genome (lysogenic pathway), where it is dormant until activated.
Mutant λ phages designed for cloning have been constructed. An especially useful one called λgt-λβ contains only two EcoRI cleavage sites instead of the five normally present (Figure 5.14). After cleavage, the middle segment of this λ DNA molecule can be removed. The two remaining pieces of DNA (called arms) have a combined length equal to 72% of a normal genome length. This amount of DNA is too little to be packaged into a λ particle, which can take up only DNA measuring from 78% to 105% of a normal genome. However, a suitably long DNA insert (such as 10 kb) between the two ends of λ DNA enables such a recombinant DNA molecule (93% of normal length) to be packaged. Nearly all infectious λ particles formed in this way will contain an inserted piece of foreign DNA. Another advantage of using these modified viruses as vectors is that they enter bacteria much more easily than do plasmids. Among the variety of λ mutants that have been constructed for use as cloning vectors, one of them, called a cosmid, is essentially a hybrid of λ phage and a plasmid that can serve as a vector for large DNA inserts (as large as 45 kb).
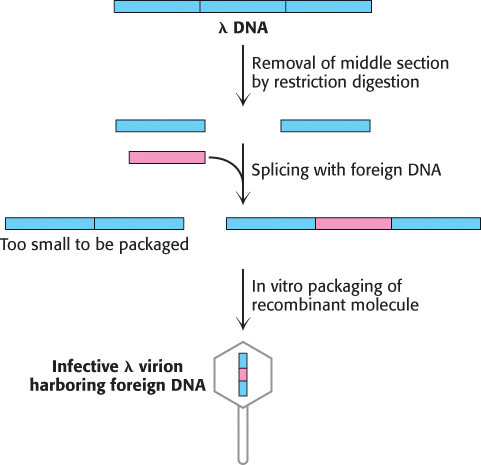
FIGURE 5.14Mutant λ phage as a cloning vector. The packaging process selects DNA molecules that contain an insert (colored red). DNA molecules that have resealed without an insert are too small to be efficiently packaged.
Bacterial and yeast artificial chromosomes
Much larger pieces of DNA can be propagated in bacterial artificial chromosomes (BACs) or yeast artificial chromosomes (YACs). BACs are highly engineered versions of the E. coli fertility (F) factor that can include inserts as large as 300 kb. YACs contain a centromere, an autonomously replicating sequence (ARS, where replication begins), a pair of telomeres (normal ends of eukaryotic chromosomes), selectable marker genes, and a cloning site (Figure 5.15). Inserts as large as 1000 kb can be cloned into YAC vectors.
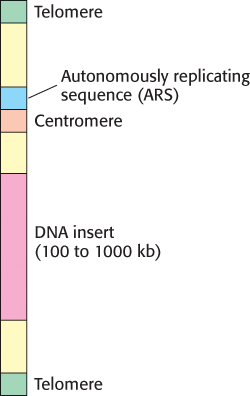
FIGURE 5.15Diagram of a yeast artificial chromosome (YAC). These vectors include features necessary for replication and stability in yeast cells.
Specific genes can be cloned from digests of genomic DNA
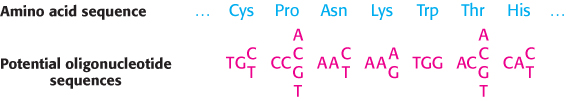
FIGURE 5.16Probes generated from a protein sequence. A probe can be generated by synthesizing all possible oligonucleotides encoding a particular sequence of amino acids. Because of the degeneracy of the genetic code, 256 distinct oligonucleotides must be synthesized to ensure that the probe matching the sequence of seven amino acids in this example is present.
Ingenious cloning and selection methods have made it possible to isolate small stretches of DNA in a genome containing more than 3 × 106 kb. The approach is to prepare a large collection (library) of DNA fragments and then to identify those members of the collection that have the gene of interest. Hence, to clone a gene that is present just once in an entire genome, two critical components must be available: a specific oligonucleotide probe for the gene of interest and a DNA library that can be screened rapidly.
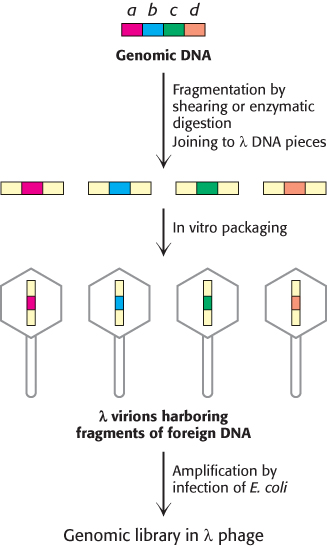
FIGURE 5.17Creation of a genomic library. A genomic library can be created from a digest of a whole complex genome. After fragmentation of the genomic DNA into overlapping segments, the DNA is inserted into the λ phage vector (shown in yellow). Packaging into virions and amplification by infection in E. coli yields a genomic library.
How is a specific probe obtained? In one approach, a probe for a gene can be prepared if a part of the amino acid sequence of the protein encoded by the gene is known. Peptide sequencing of a purified protein (Chapter 3) or knowledge of the sequence of a homologous protein from a related species (Chapter 6) are two potential sources of such information. However, a problem arises because a single peptide sequence can be encoded by a number of different oligonucleotides (Figure 5.16). Thus, for this purpose, peptide sequences containing tryptophan and methionine are preferred, because these amino acids are specified by a single codon, whereas other amino acid residues have between two and six codons (Table 4.5). All the possible DNA sequences (or their complements) that encode the targeted peptide sequence are synthesized by the solid-phase method and made radioactive by phosphorylating their 5′ ends with 32P.
To prepare the DNA library, a sample containing many copies of total genomic DNA is first mechanically sheared or partly digested by restriction enzymes into large fragments (Figure 5.17). This process yields a nearly random population of overlapping DNA fragments. These fragments are then separated by gel electrophoresis to isolate the set of all fragments that are about 15 kb long. Synthetic linkers are attached to the ends of these fragments, cohesive ends are formed, and the fragments are then inserted into a vector, such as λ phage DNA, prepared with the same cohesive ends. E. coli bacteria are then infected with these recombinant phages. These phages replicate themselves and then lyse their bacterial hosts. The resulting lysate contains fragments of human DNA housed in a sufficiently large number of virus particles to ensure that nearly the entire genome is represented. These phages constitute a genomic library. Phages can be propagated indefinitely such that the library can be used repeatedly over long periods.
This genomic library is then screened to find the very small number of phages harboring the gene of interest. For the human genome, a calculation shows that a 99% probability of success requires screening about 500,000 clones; hence, a very rapid and efficient screening process is essential. Rapid screening can be accomplished by DNA hybridization.
A dilute suspension of the recombinant phages is first plated on a lawn of bacteria (Figure 5.18). Where each phage particle has landed and infected a bacterium, a plaque containing identical phages develops on the plate. A replica of this master plate is then made by applying a sheet of nitrocellulose. Infected bacteria and phage DNA released from lysed cells adhere to the sheet in a pattern of spots corresponding to the plaques. Intact bacteria on this sheet are lysed with NaOH, which also serves to denature the DNA so that it becomes accessible for hybridization with a 32P-labeled probe. The presence of a specific DNA sequence in a single spot on the replica can be detected by using a radioactive complementary DNA or RNA molecule as a probe. Autoradiography then reveals the positions of spots harboring recombinant DNA. The corresponding plaques are picked out of the intact master plate and grown. A single investigator can readily screen a million clones in a day. This method makes it possible to isolate virtually any gene, provided that a probe is available.

FIGURE 5.18Screening a genomic library for a specific gene. Here, a plate is tested for plaques containing gene a of Figure 5.17.
Complementary DNA prepared from mRNA can be expressed in host cells
The preparation of eukaryotic DNA libraries presents unique challenges, especially if the researcher is interested primarily in the protein-coding region of a particular gene. Recall that most mammalian genes are mosaics of introns and exons. These interrupted genes cannot be expressed by bacteria, which lack the machinery to splice introns out of the primary transcript. However, this difficulty can be circumvented by causing bacteria to take up recombinant DNA that is complementary to mRNA, where the intronic sequences have been removed.
The key to forming complementary DNA is the enzyme reverse transcriptase. As discussed in Section 4.3, a retrovirus uses this enzyme to form a DNA–RNA hybrid in replicating its genomic RNA. Reverse transcriptase synthesizes a DNA strand complementary to an RNA template if the transcriptase is provided with a DNA primer that is base-paired to the RNA and contains a free 3′-OH group. We can use a simple sequence of linked thymidine [oligo(T)] residues as the primer. This oligo(T) sequence pairs with the poly(A) sequence at the 3′ end of most eukaryotic mRNA molecules (Section 4.4), as shown in Figure 5.19. The reverse transcriptase then synthesizes the rest of the cDNA strand in the presence of the four deoxyribonucleoside triphosphates (step 1). The RNA strand of this RNA–DNA hybrid is subsequently hydrolyzed by raising the pH (step 2). Unlike RNA, DNA is resistant to alkaline hydrolysis. The single-stranded DNA is converted into double-stranded DNA by creating another primer site. The enzyme terminal transferase adds nucleotides—for instance, several residues of dG—to the 3′ end of DNA (step 3). Oligo(dC) can bind to dG residues and prime the synthesis of the second DNA strand (step 4). Synthetic linkers can be added to this double-helical DNA for ligation to a suitable vector. Complementary DNA for all mRNA that a cell contains can be made, inserted into vectors, and then inserted into bacteria. Such a collection is called a cDNA library.
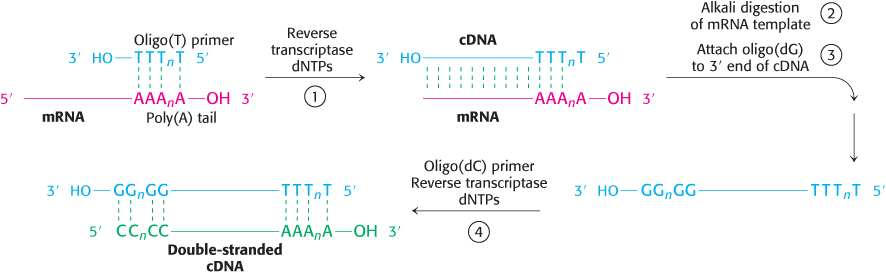
FIGURE 5.19Formation of a cDNA duplex. A complementary DNA (cDNA) duplex is created from mRNA by (1) use of reverse transcriptase to synthesize a cDNA strand, (2) digestion of the original RNA strand, (3) addition of several G bases to the DNA by terminal transferase, and (4) synthesis of a complementary DNA strand using the newly synthesized cDNA strand as a template.
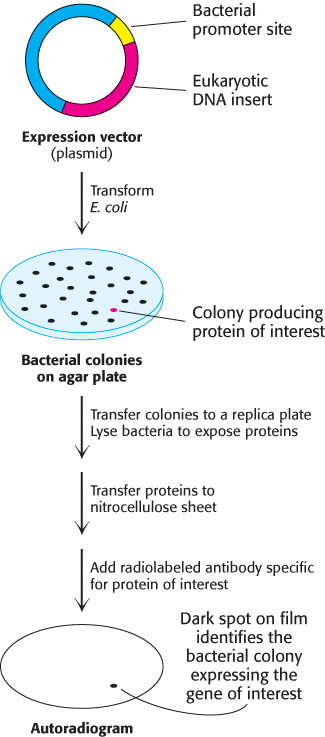
FIGURE 5.20Screening of cDNA clones. A method of screening for cDNA clones is to identify expressed products by staining with specific antibody.
Complementary DNA molecules can be inserted into expression vectors to enable the production of the corresponding protein of interest. Clones of cDNA can be screened on the basis of their capacity to direct the synthesis of a foreign protein in bacteria, a technique referred to as expression cloning. A labeled antibody specific for the protein of interest can be used to identify colonies of bacteria that express the corresponding protein product (Figure 5.20). As described earlier, spots of bacteria on a replica plate are lysed to release proteins, which bind to an applied nitrocellulose filter. With the addition of labeled antibody specific for the protein of interest, the location of the desired colonies on the master plate can be readily identified. This immunochemical screening approach can be used whenever a protein is expressed and corresponding antibody is available.
Complementary DNA has many applications beyond the generation of genetic libraries. The overproduction and purification of most eukaryotic proteins in prokaryotic cells necessitates the insertion of cDNA into plasmid vectors. For example, proinsulin, a precursor of insulin, is synthesized by bacteria-harboring plasmids that contain DNA complementary to mRNA for proinsulin (Figure 5.21). Indeed, bacteria produce much of the insulin used today by millions of diabetics.
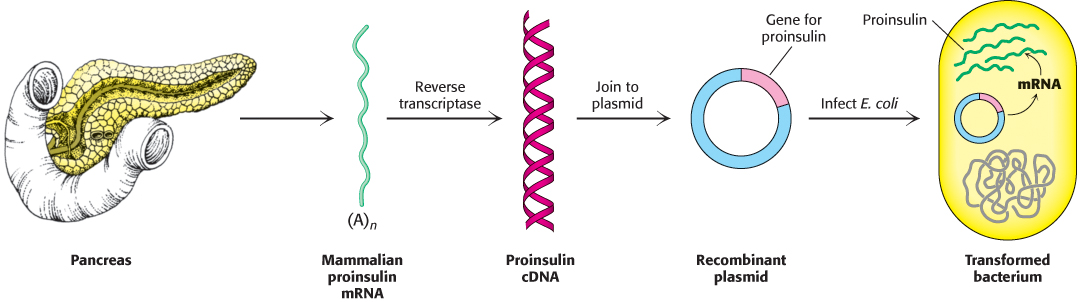
FIGURE 5.21Synthesis of proinsulin by bacteria. Proinsulin, a precursor of insulin, can be synthesized by transformed (genetically altered) clones of E. coli. The clones contain the mammalian proinsulin gene.
Proteins with new functions can be created through directed changes in DNA
Much has been learned about genes and proteins by analyzing the effects that mutations have on their structure and function. In the classic genetic approach, mutations are generated randomly throughout the genome of a host organism, and those individuals exhibiting a phenotype of interest are selected. Analysis of these mutants then reveals which genes are altered, and DNA sequencing identifies the precise nature of the changes. Recombinant DNA technology makes the creation of specific mutations feasible in vitro. We can construct new genes with designed properties by making three kinds of directed changes: deletions, insertions, and substitutions. A variety of methods can be used to introduce these types of mutations, including the following examples.
Site-directed mutagenesis.
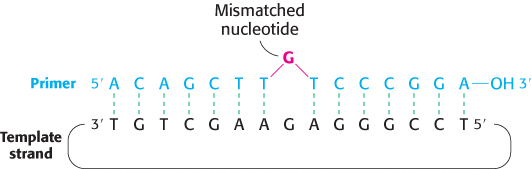
FIGURE 5.22Oligonucleotide-directed mutagenesis. A primer containing a mismatched nucleotide is used to produce a desired change in the DNA sequence.
Mutant proteins with single amino acid substitutions can be readily produced by site-directed mutagenesis (Figure 5.22). Suppose that we want to replace a particular serine residue with cysteine. This mutation can be made if (1) we have a plasmid containing the gene or cDNA for the protein and (2) we know the base sequence around the site to be altered. If the serine of interest is encoded by TCT, mutation of the central base from C to G yields the TGT codon, which encodes cysteine. This type of mutation is called a point mutation because only one base is altered. To introduce this mutation into our plasmid, we prepare an oligonucleotide primer that is complementary to this region of the gene except that it contains TGT instead of TCT. The two strands of the plasmid are separated, and the primer is then annealed to the complementary strand. The mismatch of 1 of 15 base pairs is tolerable if the annealing is carried out at an appropriate temperature. After annealing to the complementary strand, the primer is elongated by DNA polymerase, and the double-stranded circle is closed by adding DNA ligase. Subsequent replication of this duplex yields two kinds of progeny plasmid, half with the original TCT sequence and half with the mutant TGT sequence. Expression of the plasmid containing the new TGT sequence will produce a protein with the desired substitution of cysteine for serine at a unique site. We will encounter many examples of the use of site-directed mutagenesis to precisely alter regulatory regions of genes and to produce proteins with tailor-made features.
Cassette mutagenesis.
In cassette mutagenesis, a variety of mutations, including insertions, deletions, and multiple point mutations, can be introduced into the gene of interest. A plasmid harboring the original gene is cut with a pair of restriction enzymes to remove a short segment (Figure 5.23). A synthetic double-stranded oligonucleotide—the cassette—carrying the genetic alterations of interest is prepared with cohesive ends that are complementary to the ends of the cut plasmid. Ligation of the cassette into the plasmid yields the desired mutated gene product.
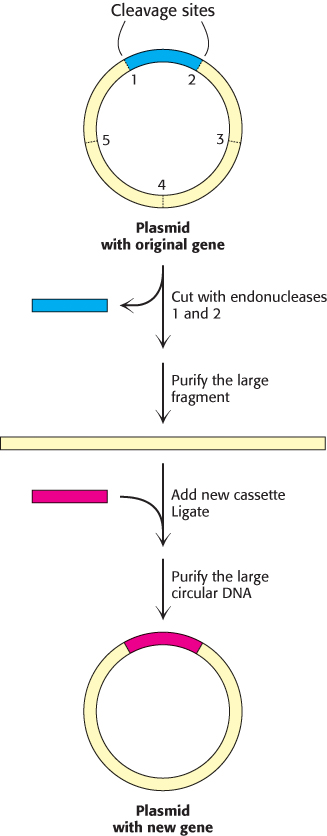
FIGURE 5.23Cassette mutagenesis. DNA is cleaved at a pair of unique restriction sites by two different restriction endonucleases. A synthetic oligonucleotide with ends that are complementary to these sites (the cassette) is then ligated to the cleaved DNA. The method is highly versatile because the inserted DNA can have any desired sequence.
Mutagenesis by PCR.
In Section 5.1, we learned how PCR can be used to amplify a specific region of DNA using primers that flank the region of interest. In fact, the creative design of PCR primers enables the introduction of specific insertions, deletions, and substitutions into the amplified sequence. A number of methods have been developed for this purpose. Here, we shall consider one: inverse PCR to introduce deletions into plasmid DNA (Figure 5.24). In this approach, primers are designed to flank the sequence to be deleted. However, these primers are oriented in the opposite direction, such that they direct the amplification of the entire plasmid, minus the region to be deleted. If each of the primers contains a 5′phosphate group, the amplified product can be recircularized with DNA ligase, yielding the desired deletion mutation.
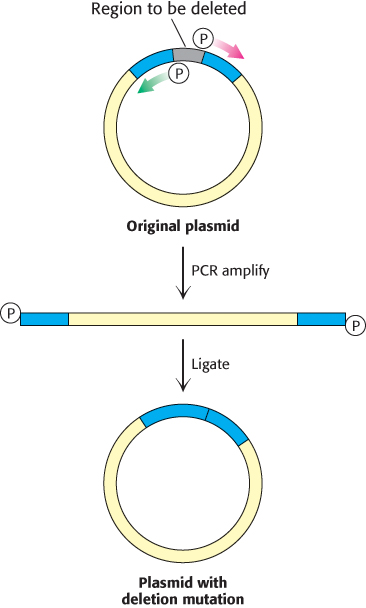
FIGURE 5.24Deletion mutagenesis by inverse PCR. A deletion can be introduced into a plasmid with primers that flank this region but are oriented away from the segment to be removed. PCR amplification yields a linear product that contains the entire plasmid minus the unwanted sequence. If the primers contained a 5’ phosphate group, this product can be recircularized using DNA ligase, generating a plasmid with the desired mutation.
Recombinant methods enable the exploration of the functional effects of disease-causing mutations
The application of recombinant DNA technology to the production of mutated proteins has had a significant effect in the study of ALS. Recall that genetic studies had identified a number of ALS-inducing mutations within the gene encoding Cu/Zn superoxide dismutase. As we shall learn in Section 18.3, SOD1 catalyzes the conversion of the superoxide radical anion into hydrogen peroxide, which, in turn, is converted into molecular oxygen and water by catalase. To study the potential effect of ALS-causing mutations on SOD1 structure and function, the SOD1 gene was isolated from a human cDNA library by PCR amplification. The amplified fragments containing the gene were then digested by an appropriate restriction enzyme and inserted into a similarly digested plasmid vector. Mutations corresponding to those observed in ALS patients were introduced into these plasmids by oligonucleotide-directed mutagenesis and the protein products were expressed and assayed for their catalytic activity. Surprisingly, these mutations did not significantly alter the enzymatic activity of the corresponding recombinant proteins. These observations have led to the prevailing notion that these mutations impart toxic properties to SOD1. Although the nature of this toxicity is not yet completely understood, one hypothesis is that mutant SOD1 is prone to form toxic aggregates in the cytoplasm of neuronal cells.