5.1The Exploration of Genes Relies on Key Tools
The Exploration of Genes Relies on Key Tools
The rapid progress in biotechnology—
1. Restriction-
2. Blotting techniques. Southern and northern blots are used to separate and identify DNA and RNA sequences, respectively. The western blot, which uses antibodies to characterize proteins, was described in Chapter 3.
3. DNA sequencing. The precise nucleotide sequence of a molecule of DNA can be determined. Sequencing has yielded a wealth of information concerning gene architecture, the control of gene expression, and protein structure.
4. Solid-
5. The polymerase chain reaction (PCR). The polymerase chain reaction leads to a billionfold amplification of a segment of DNA. One molecule of DNA can be amplified to quantities that permit characterization and manipulation. This powerful technique can be used to detect pathogens and genetic diseases, determine the source of a hair left at the scene of a crime, and resurrect genes from the fossils of extinct organisms.
A final set of techniques relies on the computer, without which, it would be impossible to catalog, access, and characterize the abundant information generated by the methods outlined above. Such uses of the computer will be presented in Chapter 6.
137
Restriction enzymes split DNA into specific fragments
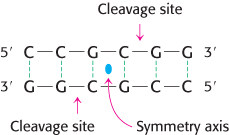
Restriction enzymes, also called restriction endonucleases, recognize specific base sequences in double-
Restriction enzymes are found in a wide variety of prokaryotes. Their biological role is to cleave foreign DNA molecules, providing the host organism with a primitive immune system. Many restriction enzymes recognize specific sequences of four to eight base pairs and hydrolyze a phosphodiester bond in each strand in this region. A striking characteristic of these cleavage sites is that they almost always possess twofold rotational symmetry. In other words, the recognized sequence is palindromic, or an inverted repeat, and the cleavage sites are symmetrically positioned. For example, the sequence recognized by a restriction enzyme from Streptomyces achromogenes is
Palindrome
A word, sentence, or verse that reads the same from right to left as it does from left to right.
Radar
Senile felines
Do geese see God?
Roma tibi subito motibus ibit amor
Derived from the Greek palindromos, “running back again.”
In each strand, the enzyme cleaves the C–
Several hundred restriction enzymes have been purified and characterized. Their names consist of a three-
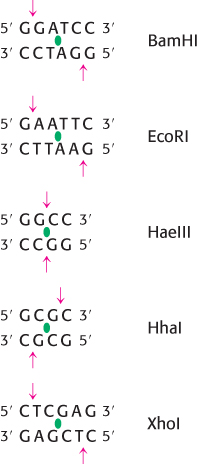
Restriction enzymes are used to cleave DNA molecules into specific fragments that are more readily analyzed and manipulated than the entire parent molecule. For example, the 5.1-
Restriction fragments can be separated by gel electrophoresis and visualized
In Chapter 3, we considered the use of gel electrophoresis to separate protein molecules (Section 3.1). Because the phosphodiester backbone of DNA is highly negatively charged, this technique is also suitable for the separation of nucleic acid fragments. Among the many applications of DNA electrophoresis are the detection of mutations that affect restriction fragment size (such as insertions and deletions) and the isolation, purification, and quantitation of a specific DNA fragment.
For most gels, the shorter the DNA fragment, the farther the migration. Polyacrylamide gels are used to separate, by size, fragments containing as many as 1000 base pairs, whereas more-
138
It is often necessary to determine if a particular base sequence is represented in a given DNA sample. For example, one may wish to confirm the presence of a specific mutation in genomic DNA isolated from patients known to be at risk for a particular disease. This specific sequence can be identified by hybridizing it with a labeled complementary DNA strand (Figure 5.3). A mixture of restriction fragments is separated by electrophoresis through an agarose gel, denatured to form single-
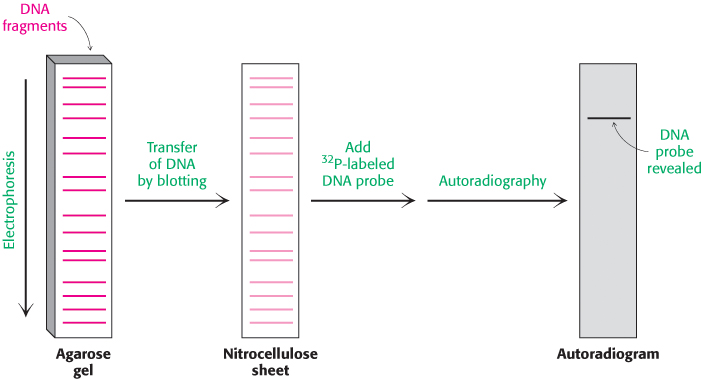
In a similar manner, RNA molecules of a specific sequence can also be readily identified. After separation by gel electrophoresis and transfer to nitrocellulose, specific sequences can be detected by DNA probes. This analogous technique for the analysis of RNA has been whimsically termed northern blotting. A further play on words accounts for the term western blotting, which refers to a technique for detecting a particular protein by staining with specific antibody (Section 3.3).
DNA can be sequenced by controlled termination of replication
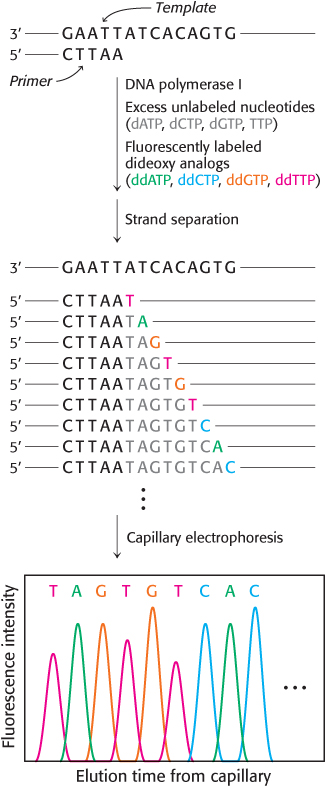
The analysis of DNA structure and its role in gene expression have been markedly facilitated by the development of powerful techniques for the sequencing of DNA molecules. One of the first and most widely-
139
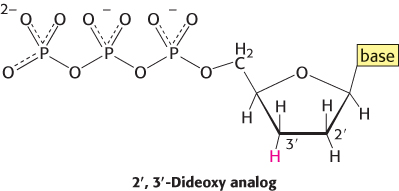
The incorporation of this analog blocks further growth of the new chain because it lacks the 3′-hydroxyl terminus needed to form the next phosphodiester bond. The concentration of the dideoxy analog is low enough that chain termination will take place only occasionally. The polymerase will insert the correct nucleotide sometimes and the dideoxy analog other times, stopping the reaction. For instance, if the dideoxy analog of dATP is present, fragments of various lengths are produced, but all will be terminated by the dideoxy analog. Importantly, this dideoxy analog of dATP will be inserted only where a T was located in the DNA being sequenced. Thus, the fragments of different length will correspond to the positions of T.
The resulting fragments are separated by a technique known as capillary electrophoresis, in which the mixture is passed through a very narrow tube containing a gel matrix at high voltage to achieve efficient separation within a short time. As the DNA fragments emerge from the capillary, they are detected by their fluorescence; the sequence of their colors directly gives the base sequence. Sequences of as many as 1000 bases can be determined in this way. Indeed, automated Sanger sequencing machines can read more than 1 million bases per day.
DNA probes and genes can be synthesized by automated solid-phase methods
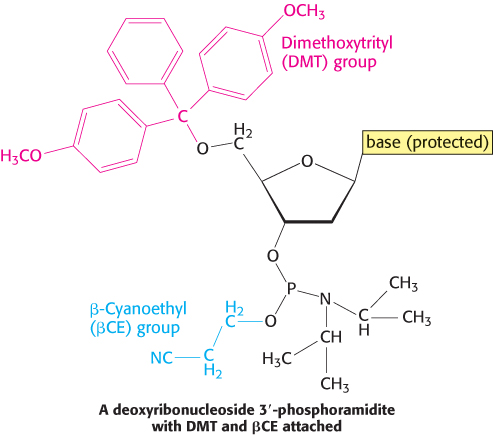
DNA strands, like polypeptides (Section 3.4), can be synthesized by the sequential addition of activated monomers to a growing chain that is linked to a solid support. The activated monomers are protected deoxyribonucleoside 3′-phosphoramidites. In step 1, the 3′-phosphorus atom of this incoming unit becomes joined to the 5′-oxygen atom of the growing chain to form a phosphite triester (Figure 5.5). The 5′-OH group of the activated monomer is unreactive because it is blocked by a dimethoxytrityl (DMT) protecting group, and the 3′-phosphoryl oxygen atom is rendered unreactive by attachment of the β-cyanoethyl (βCE) group. Likewise, amino groups on the purine and pyrimidine bases are blocked.
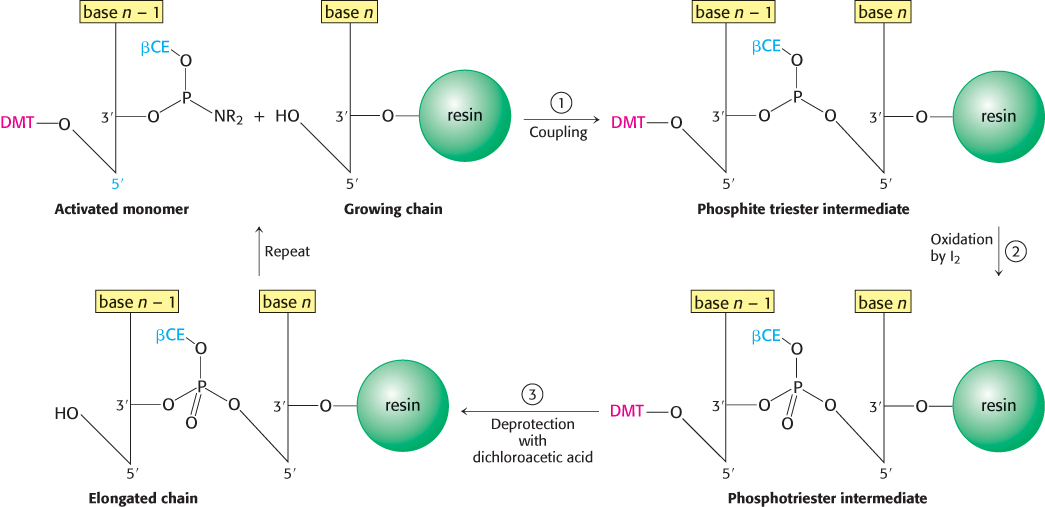
Coupling is carried out under anhydrous conditions because water reacts with phosphoramidites. In step 2, the phosphite triester (in which P is trivalent) is oxidized by iodine to form a phosphotriester (in which P is pentavalent). In step 3, the DMT protecting group on the 5′-OH group of the growing chain is removed by the addition of dichloroacetic acid, which leaves other protecting groups intact. The DNA chain is now elongated by one unit and ready for another cycle of addition. Each cycle takes only about 10 minutes and usually elongates more than 99% of the chains.
140
This solid-
The ability to rapidly synthesize DNA chains of any selected sequence opens many experimental avenues. For example, a synthesized oligonucleotide labeled at one end with 32P or a fluorescent tag can be used to search for a complementary sequence in a very long DNA molecule or even in a genome consisting of many chromosomes. The use of labeled oligonucleotides as DNA probes is powerful and general. For example, a DNA probe that can base-
141
Selected DNA sequences can be greatly amplified by the polymerase chain reaction
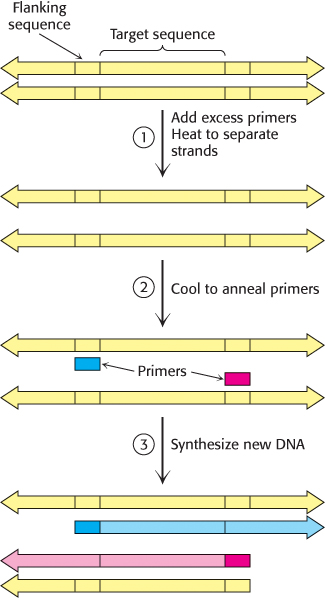
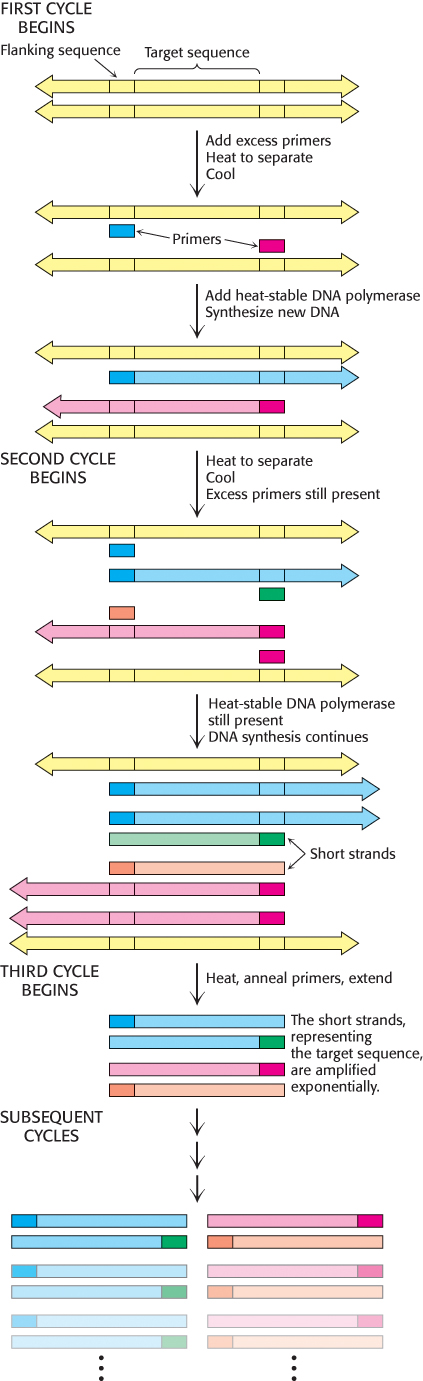
In 1984, Kary Mullis devised an ingenious method called the polymerase chain reaction (PCR) for amplifying specific DNA sequences. Consider a DNA duplex consisting of a target sequence surrounded by nontarget DNA. Millions of copies of the target sequences can be readily obtained by PCR if the sequences flanking the target are known. PCR is carried out by adding the following components to a solution containing the target sequence: (1) a pair of primers that hybridize with the flanking sequences of the target, (2) all four deoxyribonucleoside triphosphates (dNTPs), and (3) a heat-
1. Strand separation. The two strands of the parent DNA molecule are separated by heating the solution to 95°C for 15 s.
2. Hybridization of primers. The solution is then abruptly cooled to 54°C to allow each primer to hybridize to a DNA strand. One primer hybridizes to the 3′ end of the target on one strand, and the other primer hybridizes to the 3′ end on the complementary target strand. Parent DNA duplexes do not form, because the primers are present in large excess. Primers are typically from 20 to 30 nucleotides long.
3. DNA synthesis. The solution is then heated to 72°C, the optimal temperature for heat-
These three steps—
Several features of this remarkable method for amplifying DNA are noteworthy. First, the sequence of the target need not be known. All that is required is knowledge of the flanking sequences so that complementary primers can be synthesized. Second, the target can be much larger than the primers. Targets larger than 10 kb have been amplified by PCR. Third, primers do not have to be perfectly matched to flanking sequences to amplify targets. With the use of primers derived from a gene of known sequence, it is possible to search for variations on the theme. In this way, families of genes are being discovered by PCR. Fourth, PCR is highly specific because of the stringency of hybridization at relatively high temperature. Stringency is the required closeness of the match between primer and target, which can be controlled by temperature and salt. At high temperatures, only the DNA between hybridized primers is amplified. A gene constituting less than a millionth of the total DNA of a higher organism is accessible by PCR. Fifth, PCR is exquisitely sensitive. A single DNA molecule can be amplified and detected.
142
PCR is a powerful technique in medical diagnostics, forensics, and studies of molecular evolution
 PCR can provide valuable diagnostic information in medicine. Bacteria and viruses can be readily detected with the use of specific primers. For example, PCR can reveal the presence of small amounts of DNA from the human immunodeficiency virus (HIV) in persons who have not yet mounted an immune response to this pathogen. In these patients, assays designed to detect antibodies against the virus would yield a false negative test result. Finding Mycobacterium tuberculosis bacilli in tissue specimens is slow and laborious. With PCR, as few as 10 tubercle bacilli per million human cells can be readily detected. PCR is a promising method for the early detection of certain cancers. This technique can identify mutations of certain growth-
PCR can provide valuable diagnostic information in medicine. Bacteria and viruses can be readily detected with the use of specific primers. For example, PCR can reveal the presence of small amounts of DNA from the human immunodeficiency virus (HIV) in persons who have not yet mounted an immune response to this pathogen. In these patients, assays designed to detect antibodies against the virus would yield a false negative test result. Finding Mycobacterium tuberculosis bacilli in tissue specimens is slow and laborious. With PCR, as few as 10 tubercle bacilli per million human cells can be readily detected. PCR is a promising method for the early detection of certain cancers. This technique can identify mutations of certain growth-
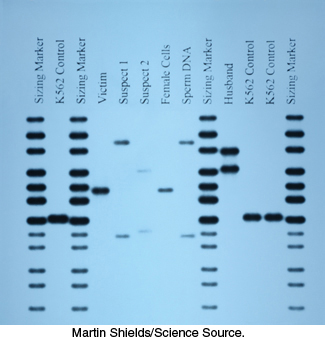
In addition, PCR has made an impact on forensics and legal medicine. An individual DNA profile is highly distinctive because many genetic loci are highly variable within a population. For example, variations at one specific location determine a person’s HLA type (human leukocyte antigen type; Section 34.5); organ transplants are rejected when the HLA types of the donor and recipient are not sufficiently matched. PCR amplification of multiple genes is being used to establish biological parentage in disputed paternity and immigration cases. Analyses of blood stains and semen samples by PCR have implicated guilt or innocence in numerous assault and rape cases (Figure 5.8). The root of a single shed hair found at a crime scene contains enough DNA for typing by PCR.
DNA is a remarkably stable molecule, particularly when shielded from air, light, and water. Under such circumstances, large fragments of DNA can remain intact for thousands of years or longer. PCR provides an ideal method for amplifying such ancient DNA molecules so that they can be detected and characterized (Section 6.5). PCR can also be used to amplify DNA from microorganisms that have not yet been isolated and cultured. As will be discussed in Chapter 6, sequences from these PCR products can be sources of considerable insight into evolutionary relationships between organisms.
143
The tools for recombinant DNA technology have been used to identify disease-causing mutations
Let us consider how the techniques just described have been utilized in concert to study ALS, introduced at the beginning of this chapter. Five percent of all patients suffering from ALS have family members who also have been diagnosed with the disease. A heritable disease pattern is indicative of a strong genetic component of disease causation. To identify these disease-
After the probable location of one disease-