5.4
Eukaryotic Genes Can Be Quantitated and Manipulated with Considerable Precision
After a gene of interest has been identified, cloned, and sequenced, it is often desirable to understand how that gene and its corresponding protein product function in the context of a whole cell or organism. It is now possible to determine how the expression of a particular gene is regulated, how mutations in the gene affect the function of the corresponding protein product, and how the behavior of an entire cell or model organism is altered by the introduction of mutations within specific genes. Levels of transcription of large families of genes within cells and tissues can be readily quantitated and compared across a range of environmental conditions. Eukaryotic genes can be introduced into bacteria, and the bacteria can be used as factories to produce a desired protein product. DNA can also be introduced into the cells of higher organisms. Genes introduced into animals are valuable tools for examining gene action, and they are the basis of gene therapy. Genes introduced into plants can make the plants resistant to pests, able to grow in harsh conditions, or carry greater quantities of essential nutrients. The manipulation of eukaryotic genes holds much promise as a source of medical and agricultural benefits.
Gene-expression levels can be comprehensively examined
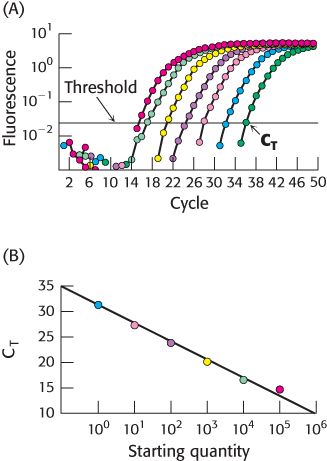
FIGURE 5.29Quantitative PCR. (A) In qPCR, fluorescence is monitored in the course of PCR amplification to determine CT, the cycle at which this signal exceeds a defined threshold. Each color represents a different starting quantity of DNA. (B) CT values are inversely proportional to the number of copies of the original cDNA template.
[Data from N. J. Walker, Science 296:557–559, 2002.]
Most genes are present in the same quantity in every cell—namely, one copy per haploid cell or two copies per diploid cell. However, the level at which a gene is expressed, as indicated by mRNA quantities, can vary widely, ranging from no expression to hundreds of mRNA copies per cell. Gene-expression patterns vary from cell type to cell type, distinguishing, for example, a muscle cell from a nerve cell. Even within the same cell, gene-expression levels may vary as the cell responds to changes in physiological circumstances. Note that mRNA levels sometimes correlate with the levels of proteins expressed, but this correlation does not always hold. Thus, care must be exercised when interpreting the results of mRNA levels alone.
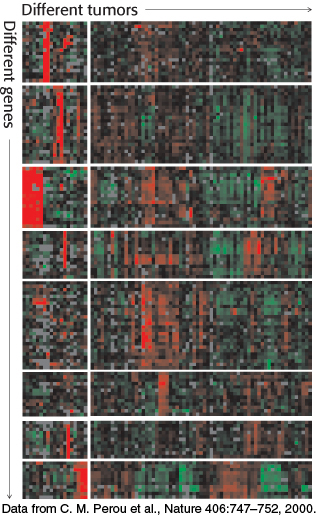
FIGURE 5.31Gene-expression analysis with the use of microarrays. The expression levels of thousands of genes can be simultaneously analyzed with DNA microarrays. Here, an analysis of 1733 genes in 84 breast tumor samples reveals that the tumors can be divided into distinct classes based on their gene-expression patterns. In this “heat map” representation, each row represents a different gene and each column represents a different breast tumor sample (i.e., a separate microarray experiment). Red corresponds to gene induction and green corresponds to gene repression.
[Data from C. M. Perou et al., Nature 406:747–752, 2000.]
The quantity of individual mRNA transcripts can be determined by quantitative PCR (qPCR), or real-time PCR. RNA is first isolated from the cell or tissue of interest. With the use of reverse transcriptase, cDNA is prepared from this RNA sample. In one qPCR approach, the transcript of interest is PCR amplified with the appropriate primers in the presence of the dye SYBR Green I, which fluoresces brightly when bound to double-stranded DNA. In the initial PCR cycles, not enough duplex is present to allow a detectable fluorescence signal. However, after repeated PCR cycles, the fluorescence intensity exceeds the detection threshold and continues to rise as the number of duplexes corresponding to the transcript of interest increases (Figure 5.29). Importantly, the cycle number at which the fluorescence becomes detectable over a defined threshold (or CT) is indirectly proportional to the number of copies of the original template. After the relation between the original copy number and the CT has been established with the use of a known standard, subsequent qPCR experiments can be used to determine the number of copies of any desired transcript in the original sample, provided the appropriate primers are available.
Although qPCR is a powerful technique for quantitation of a small number of transcripts in any given experiment, we can now use our knowledge of complete genome sequences to investigate an entire transcriptome, the pattern and level of expression of all genes in a particular cell or tissue. One of the most powerful methods for this purpose is based on hybridization. Single-stranded oligonucleotides whose sequences correspond to coding regions of the genome are affixed to a solid support such as a microscope slide, creating a DNA microarray. Importantly, the position of each sequence within the array is known. mRNA is isolated from the cells of interest (a tumor, for example) as well as a control sample (Figure 5.30). From this mRNA, cDNA is prepared (Section 5.2) in the presence of fluorescent nucleotides using different labels, usually green and red, for the two samples. The samples are combined, separated into single strands, and hybridized to the slide. The relative levels of green and red fluorescence at each spot indicate the differences in expression for each gene. DNA chips have been prepared such that thousands of transcript levels can be assessed in a single experiment. Hence, over several arrays, the differences in expression of many genes across a number of different cell types or conditions can be measured (Figure 5.31).
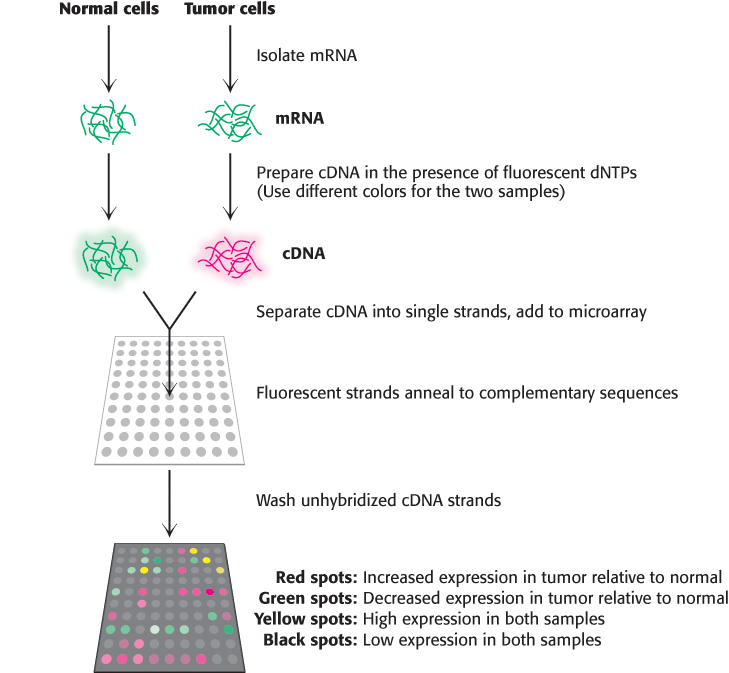
FIGURE 5.30Using DNA microarrays to measure gene expression changes in a tumor. mRNA is isolated from two samples, tumor cells and a control sample. From these transcripts, cDNA is prepared in the presence of a fluorescent nucleotide, with a red label for the tumor sample and a green label for the control sample. The cDNA strands are separated, hybridized to the microarray, and the unbound DNA is washed away. Spots that are red indicate genes which are expressed more highly in the tumor, while the green spots indicate reduced expression relative to control. Spots that are black or yellow indicate comparable expression at either low or high levels, respectively.
[Information from D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry 6th ed. (W. H. Freeman and Company, 2013)]
Microarray analyses can be quite informative in the study of gene-expression changes in diseased mammals compared with their healthy counterparts. As noted earlier, although ALS-causing mutations within the SOD1 gene had been identified, the mechanism by which the mutant SOD1 protein ultimately leads to motor-neuron loss remains a mystery. Many research groups have used microarray analysis of neuronal cells isolated from humans and mice carrying SOD1 gene mutations to search for clues into the pathways of disease progression and to suggest potential avenues for treatment. These studies have implicated a variety of biochemical pathways, including immunological activation, handling of oxidative stress, and protein degradation, in the cellular response to the mutant, toxic forms of SOD1.
New genes inserted into eukaryotic cells can be efficiently expressed
Bacteria are ideal hosts for the amplification of DNA molecules. They can also serve as factories for the production of a wide range of prokaryotic and eukaryotic proteins. However, bacteria lack the necessary enzymes to carry out posttranslational modifications such as the specific cleavage of polypeptides and the attachment of carbohydrate units. Thus, many eukaryotic genes can be expressed correctly only in eukaryotic host cells. The introduction of recombinant DNA molecules into cells of higher organisms can also be a source of insight into how their genes are organized and expressed. How are genes turned on and off in embryological development? How does a fertilized egg give rise to an organism with highly differentiated cells that are organized in space and time? These central questions of biology can now be fruitfully approached by expressing foreign genes in mammalian cells.
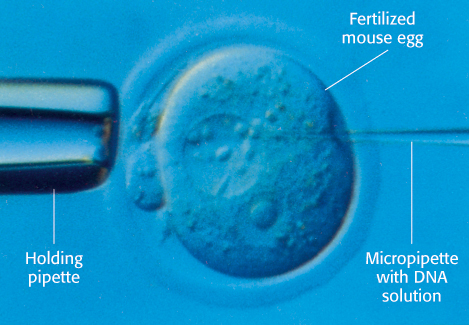
FIGURE 5.32Microinjection of DNA. Cloned plasmid DNA is being microinjected into the male pronucleus of a fertilized mouse egg.
Recombinant DNA molecules can be introduced into animal cells in several ways. In one method, foreign DNA molecules precipitated by calcium phosphate are taken up by animal cells. A small fraction of the imported DNA becomes stably integrated into the chromosomal DNA. The efficiency of incorporation is low, but the method is useful because it is easy to apply. In another method, DNA is microinjected into cells. A fine-tipped glass micropipette containing a solution of foreign DNA is inserted into a nucleus (Figure 5.32). A skilled investigator can inject hundreds of cells per hour. About 2% of injected mouse cells are viable and contain the new gene. In a third method, viruses are used to introduce new genes into animal cells. The most effective vectors are retroviruses, whose genomes are encoded by RNA and replicate through DNA intermediates. A striking feature of the life cycle of a retrovirus is that the double-helical DNA form of its genome, produced by the action of reverse transcriptase, becomes randomly incorporated into host chromosomal DNA. This DNA version of the viral genome, called proviral DNA, can be efficiently expressed by the host cell and replicated along with normal cellular DNA. Retroviruses do not usually kill their hosts. Foreign genes have been efficiently introduced into mammalian cells by infecting them with vectors derived from the Moloney murine leukemia virus, a retrovirus which can accept inserts as long as 6 kb. Some genes introduced by this vector into the genome of a transformed host cell are efficiently expressed.
Two other viral vectors are extensively used. Vaccinia virus, a large DNA-containing virus, replicates in the cytoplasm of mammalian cells, where it shuts down host-cell protein synthesis. Baculovirus infects insect cells, which can be conveniently cultured. Insect larvae infected with this virus can serve as efficient protein factories. Vectors based on these large-genome viruses have been engineered to express DNA inserts efficiently.
Transgenic animals harbor and express genes introduced into their germ lines
As shown in Figure 5.32, plasmids harboring foreign genes can be microinjected into the male pronucleus of fertilized mouse eggs, which are then inserted into the uterus of a foster-mother mouse. A subset of the resulting embryos in this host will then harbor the foreign gene; these embryos may develop into mature animals. Southern blotting or PCR analysis of DNA isolated from the progeny can be used to determine which offspring carry the introduced gene. These transgenic mice are a powerful means of exploring the role of a specific gene in the development, growth, and behavior of an entire organism. Transgenic animals often serve as useful models for a particular disease process, enabling researchers to test the efficacy and safety of a newly developed therapy.
Let us return to our example of ALS. Research groups have generated transgenic mouse lines that express forms of human superoxide dismutase that harbor mutations matching those identified in earlier genetic analyses. Many of these strains exhibit a clinical picture similar to that observed in ALS patients: progressive weakness of voluntary muscles and eventual paralysis, motor-neuron loss, and rapid progression to death. Since their first characterization in 1994, these strains continue to serve as valuable sources of information for the exploration of the mechanism, and potential treatment, of ALS.
Gene disruption and genome editing provide clues to gene function and opportunities for new therapies
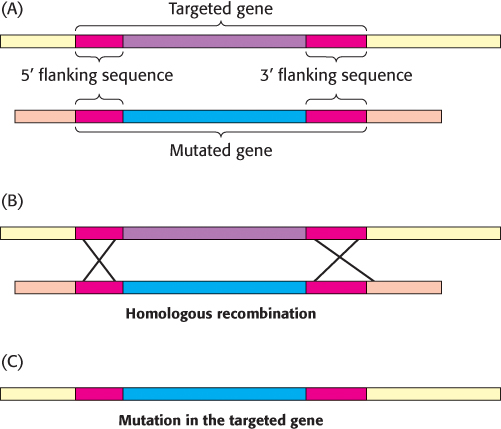
FIGURE 5.33Gene disruption by homologous recombination. (A) A mutated version of the gene to be disrupted is constructed, maintaining some regions of homology with the normal gene (red). When the foreign mutated gene is introduced into an embryonic stem cell, (B) recombination takes place at regions of homology and (C) the normal (targeted) gene is replaced, or “knocked out,” by the foreign gene. The cell is inserted into embryos, and mice lacking the gene (knockout mice) are produced.
The function of a gene can also be probed by inactivating it and looking for resulting abnormalities. Powerful methods have been developed for accomplishing gene disruption (also called gene knockout) in organisms such as yeast and mice. These methods rely on the process of homologous recombination (Section 28.5), in which two DNA molecules with strong sequence similarity exchange segments. If a region of foreign DNA is flanked by sequences that have high homology to a particular region of genomic DNA, two recombination events will yield the transfer of the foreign DNA into the genome (Figure 5.33). In this manner, specific genes can be targeted if their flanking nucleotide sequences are known.
For example, the gene-knockout approach has been applied to the genes encoding gene-regulatory proteins (also called transcription factors) that control the differentiation of muscle cells. When both copies of the gene for the regulatory protein myogenin are disrupted, an animal dies at birth because it lacks functional skeletal muscle. Microscopic inspection reveals that the tissues from which muscle normally forms contain precursor cells that have failed to differentiate fully (Figure 5.34A and B). Heterozygous mice containing one normal myogenin gene and one disrupted gene appear normal, suggesting that a reduced level of gene expression is still sufficient for normal muscle development. The generation and characterization of this knockout strain provided strong evidence that functional myogenin is essential for proper development of skeletal muscle tissue (Figure 5.34C). Analogous studies have probed the function of many other genes to generate animal models for known human genetic diseases.
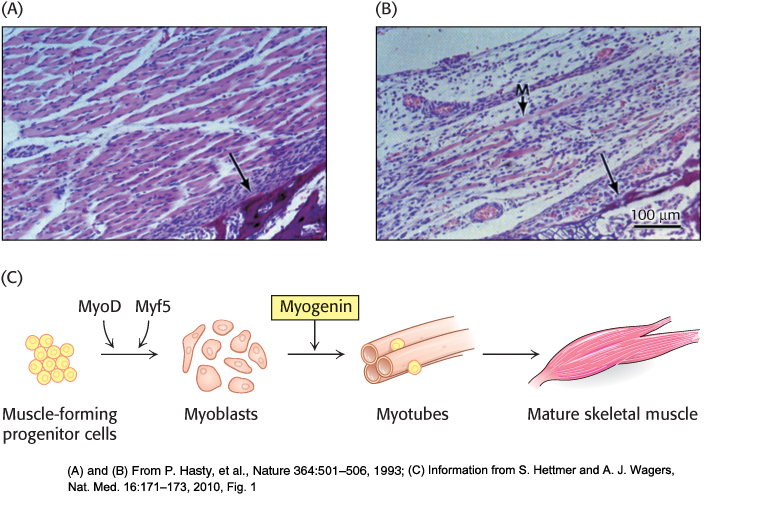
FIGURE 5.34Consequences of gene disruption. Sections of muscle from normal (A) and myogenin-knockout (B) mice, as viewed under the light microscope. The unlabeled arrows in both panels identify comparable sections of the pelvic bone, indicating that similar anatomical regions are depicted. Muscles do not develop properly in mice having both myogenin genes disrupted. A poorly formed muscle fiber in the knockout strain is indicated by the M arrow. (C) The development of mature skeletal muscle from progenitor cells is a highly regulated process involving a number of intermediate cell types and multiple transcription factors. Through the gene-disruption studies in (A) and (B), myogenin was identified as an essential component of this pathway.
[(A) and (B) From P. Hasty, et al., Nature 364:501–506, 1993; (C) Information from S. Hettmer and A. J. Wagers, Nat. Med. 16:171–173, 2010, Fig. 1]
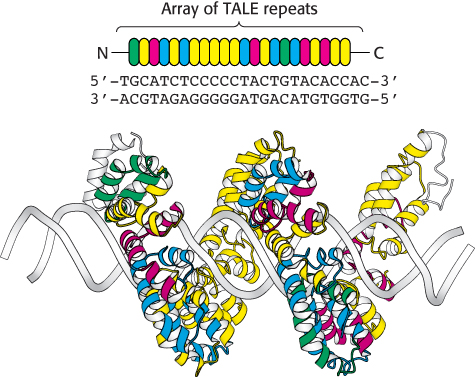
FIGURE 5.35TALE repeats recognize individual bases in DNA. Each TALE repeat contains 34 amino acids, two of which specify its nucleotide binding partner. In this figure, the identity of these residues is indicated by the color of the repeat. TALE proteins can be designed to uniquely recognize extended oligonucleotide sequences. In this example, a 22 base-pair sequence is bound by a single TALE protein, the bacterial effector PthXo1.
[Drawn from 3UGM.pdb]
Manipulation of genomic DNA using homologous recombination, while a powerful tool, has limitations. Introduction of point mutations into genes, rather than knocking out the entire gene, can be difficult and time-consuming. In addition, these methods are generally limited to specific model organisms, such as yeast, mice, and fruit flies. Over the past 10 years, new methods for the highly specific modification of genomic DNA, or genome editing, have emerged. These approaches rely on engineered site-specific nucleases that introduce double-strand breaks at precisely-determined sequences within genomic DNA. In one approach, the nonspecific nuclease domain of the restriction enzyme FokI is fused to a DNA-binding domain designed to bind to a particular DNA sequence. In zinc-finger nucleases (ZFNs), the DNA-binding domain contains a series of zinc finger domains (Section 32.2), small zinc-binding motifs that each recognize a sequence of three base pairs. The preferred DNA binding sequence can be altered by changing the identity of only four contact residues within each finger. In transcription activator-like effector nucleases (TALENs), the DNA-binding domain is comprised of an array of TALE repeats. Each repeat contains 34 amino acids and two α-helices, yet only two of these residues (at positions 12 and 13) are responsible for the unique recognition of a single nucleotide within the double helix (Figure 5.35). Mutation of these residues within an array of repeats enables the recognition of a vast number of possible DNA target sequences with a high degree of specificity.
How do these engineered nucleases effect a change in the genomic DNA sequence? Upon binding of the ZFN or TALEN to DNA, the nuclease domain cleaves the phosphate backbone of one of the DNA strands. A second nuclease, designed to recognize the opposite strand, introduces a second cleavage site, yielding a complete double-stranded break. The resulting cleavage site is repaired by the DNA repair machinery of the host cell (Section 28.5). If a DNA fragment containing the desired sequence change is simultaneously introduced with the nucleases, the repair machinery will use this donor template to introduce these changes directly into the genomic sequence, in a manner similar to the homologous recombination process described above (Figure 5.36).
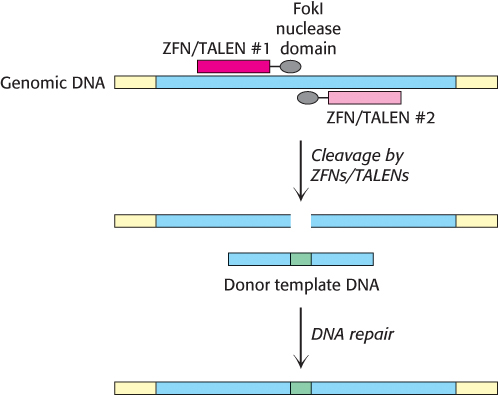
FIGURE 5.36Genome editing by site-specific nucleases. A pair of ZFNs or TALENs cleave opposite strands of a targeted gene (blue) within the genome. The DNA repair machinery of the cell will use a homologous donor template DNA fragment to fix the double-strand break, incorporating the desired modifications (green) into the targeted gene.
Site-specific nuclease-based genome editing methods have now been applied to a variety of species, including model organisms used in the laboratory (rat, zebrafish, and fruit fly), various forms of livestock (pig, cow), and a number of plants. In addition, their use as therapeutic tools in humans is currently under investigation. For example, a ZFN which inactivates the human CCR5 gene, a coreceptor for cellular invasion of human immunodeficiency virus (HIV), is currently in clinical trials for the treatment of patients infected with HIV.
RNA interference provides an additional tool for disrupting gene expression
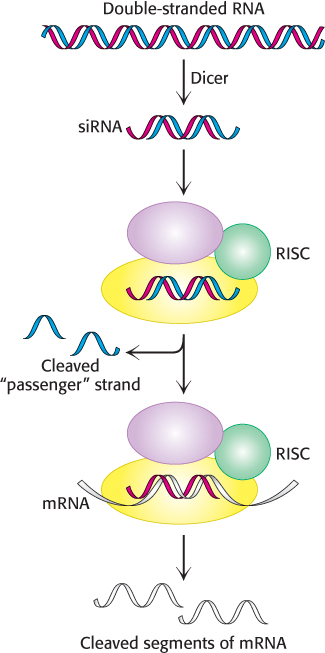
FIGURE 5.37RNA interference mechanism. A double-stranded RNA molecule is cleaved into 21-bp fragments by the enzyme Dicer to produce siRNAs. These siRNAs are incorporated into the RNA-induced silencing complex (RISC), where the single-stranded RNAs guide the cleavage of mRNAs that contain complementary sequences.
An extremely powerful tool for disrupting gene expression was serendipitously discovered in the course of studies that required the introduction of RNA into a cell. The introduction of a specific double-stranded RNA molecule into a cell was found to suppress the transcription of genes that contained sequences present in the double-stranded RNA molecule. Thus, the introduction of a specific RNA molecule can interfere with the expression of a specific gene.
The mechanism of RNA interference has been largely established (Figure 5.37). When a double-stranded RNA molecule is introduced into an appropriate cell, the RNA is cleaved by the enzyme Dicer into fragments approximately 21 nucleotides in length. Each fragment, termed a small interfering RNA (siRNA), consists of 19 bp of double-stranded RNA and 2 bases of unpaired RNA on each 5′ end. The siRNA is loaded into an assembly of several proteins referred to as the RNA-induced silencing complex (RISC), which unwinds the RNA duplex and cleaves one of the strands, the so-called passenger strand. The uncleaved single-stranded RNA segment, the guide strand, remains incorporated into the enzyme. The fully assembled RISC cleaves mRNA molecules that contain exact complements of the guide-strand sequence. Thus, levels of such mRNA molecules are dramatically reduced. The technique of RNA interference is called gene knockdown, because the expression of the gene is reduced but not eliminated, as is the case with gene knockouts.
The machinery necessary for RNA interference is found in many cells. In some organisms such as C. elegans, RNA interference is quite efficient. Indeed, RNA interference can be induced simply by feeding C. elegans strains of E. coli that have been engineered to produce appropriate double-stranded RNA molecules. Although not as efficient in mammalian cells, RNA interference has emerged as a powerful research tool for reducing the expression of specific genes. Moreover, initial clinical trials of therapies based on RNA interference are underway.
Tumor-inducing plasmids can be used to introduce new genes into plant cells

FIGURE 5.38Tumors in plants. Crown gall, a plant tumor, is caused by a bacterium (Agrobacterium tumefaciens) that carries a tumor-inducing plasmid (Ti plasmid).
[From M. Escobar et al., Proc. Natl. Acad. Sci. U. S. A. 98:13437–13442, 2001. Copyright © 2001 National Academy of Sciences, U. S. A.]
The common soil bacterium Agrobacterium tumefaciens infects plants and introduces foreign genes into plant cells (Figure 5.38). A lump of tumor tissue called a crown gall grows at the site of infection. Crown galls synthesize opines, a group of amino acid derivatives that are metabolized by the infecting bacteria. In essence, the metabolism of the plant cell is diverted to satisfy the highly distinctive appetite of the intruder. Tumor-inducing plasmids (Ti plasmids) that are carried by A. tumefaciens carry instructions for the switch to the tumor state and the synthesis of opines. A small part of the Ti plasmid becomes integrated into the genome of infected plant cells; this 20-kb segment is called T-DNA (transferred DNA; Figure 5.39).
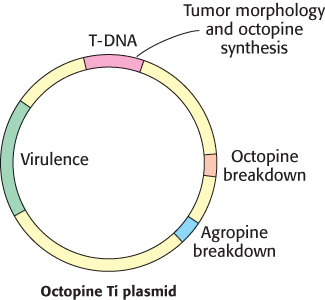
FIGURE 5.39Ti plasmids. Agrobacteria containing Ti plasmids can deliver foreign genes into some plant cells.
[Information from M. Chilton. A vector for introducing new genes into plants. Copyright © 1983 by Scientific American, Inc. All rights reserved.]
Ti-plasmid derivatives can be used as vectors to deliver foreign genes into plant cells. First, a segment of foreign DNA is inserted into the T-DNA region of a small plasmid through the use of restriction enzymes and ligases. This synthetic plasmid is added to A. tumefaciens colonies harboring naturally occurring Ti plasmids. By recombination, Ti plasmids containing the foreign gene are formed. These Ti vectors hold great promise as tools for exploring the genomes of plant cells and modifying plants to improve their agricultural value and crop yield. However, they are not suitable for transforming all types of plants. Ti-plasmid transfer is effective with dicots (broad-leaved plants such as grapes) and a few kinds of monocots but not as effective with economically important cereal monocots.
Foreign DNA can be introduced into cereal monocots as well as dicots by applying intense electric fields, a technique called electroporation (Figure 5.40). First, the cellulose wall surrounding plant cells is removed by adding cellulase; this treatment produces protoplasts, plant cells with exposed plasma membranes. Electric pulses are then applied to a suspension of protoplasts and plasmid DNA. Because high electric fields make membranes transiently permeable to large molecules, plasmid DNA molecules enter the cells. The cell wall is then allowed to reform, and the plant cells are again viable. Maize cells and carrot cells have been stably transformed in this way with the use of plasmid DNA that includes genes for resistance to herbicides. Moreover, the transformed cells efficiently express the plasmid DNA. Electroporation is also an effective means of delivering foreign DNA into animal cells and bacterial cells.
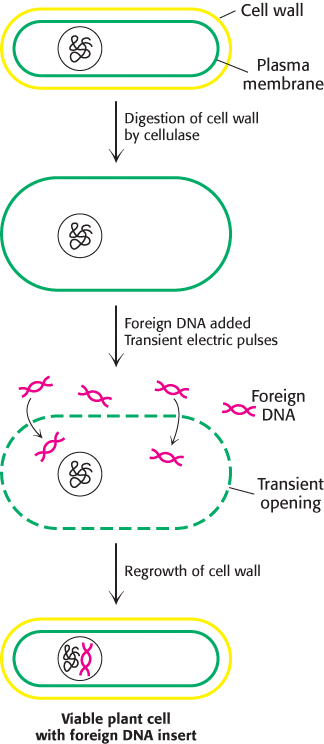
FIGURE 5.40Electroporation. Foreign DNA can be introduced into plant cells by electroporation, the application of intense electric fields to make their plasma membranes transiently permeable.
The most effective means of transforming plant cells is through the use of “gene guns,” or bombardment-mediated transformation. DNA is coated onto 1-μm-diameter tungsten pellets, and these microprojectiles are fired at the target cells with a velocity greater than 400 m s–1. Despite its apparent crudeness, this technique is proving to be the most effective way of transforming plants, especially important crop species such as soybean, corn, wheat, and rice. The gene-gun technique affords an opportunity to develop genetically modified organisms (GMOs) with beneficial characteristics, such as the ability to grow in poor soils, resistance to natural climatic variation, resistance to pests, and the ability to fortify nutritional content. These crops might be most useful in developing countries. However, the use of GMOs is highly controversial, as some fear that their safety risks have not been adequately addressed.
The first GMO to come to market was a tomato characterized by delayed ripening, rendering it ideal for shipment. Pectin is a polysaccharide that gives tomatoes their firmness and is naturally destroyed by the enzyme polygalacturonase. As pectin is destroyed, the tomatoes soften, making shipment difficult. DNA was introduced that disrupts the polygalacturonase gene. Less of the enzyme was produced, and the tomatoes stayed fresh longer. However, the tomato’s poor taste hindered its commercial success. An especially successful result of the use of Ti plasmid to modify crops is golden rice. Golden rice is a variety of genetically modified rice that contains the genes for β-carotene synthesis, a required precursor for vitamin A synthesis in humans. Consumption of this rice will benefit children and pregnant woman in parts of the world where rice is a dietary staple and vitamin A deficiency is common.
 The field of gene therapy attempts to express specific genes within the human body in such a way that beneficial results are obtained. The gene targeted for expression may be already present or specially introduced. Alternatively, gene therapy may attempt to modify genes containing sequence variations that have harmful consequences. A tremendous amount of research remains to be done before gene therapy becomes practical. Nonetheless, considerable progress has been made. For example, some people lack functional genes for adenosine deaminase and succumb to infections if exposed to a normal environment, a condition called severe combined immunodeficiency (SCID). Functional genes for this enzyme have been introduced by using gene-
The field of gene therapy attempts to express specific genes within the human body in such a way that beneficial results are obtained. The gene targeted for expression may be already present or specially introduced. Alternatively, gene therapy may attempt to modify genes containing sequence variations that have harmful consequences. A tremendous amount of research remains to be done before gene therapy becomes practical. Nonetheless, considerable progress has been made. For example, some people lack functional genes for adenosine deaminase and succumb to infections if exposed to a normal environment, a condition called severe combined immunodeficiency (SCID). Functional genes for this enzyme have been introduced by using gene-