4.4 PROTEIN FOLDING
The structures of more than 50,000 proteins have been solved, and in all cases the polypeptide backbone folds to adopt a particular conformation, a process known as protein folding. Decades have passed since the classic studies by Christian Anfinsen showing that the amino acid sequence determines the folding pattern of a protein. He showed that ribonuclease that had been completely unfolded in a denaturing solution could rapidly fold into a biologically active protein after removal of the denaturant. Ribonuclease contains eight Cys residues that form four intrachain disulfide bonds. Amazingly, on refolding, all four disulfide links formed in the correct places, even though there are 105 possible combinations. The results suggested that other, weak interactions direct protein folding and precisely position the Cys residues prior to disulfide bond formation.
Not all proteins renature as easily as ribonuclease, and the exact process by which most proteins fold is still unknown. Furthermore, some proteins require the assistance of other proteins for proper folding. Even when correctly folded, a protein structure is constantly in flux, and all of its atoms and structural elements vibrate rapidly. This inherent flexibility is essential to protein function—to achieve, for instance, a different thermodynamic state when substrate is converted to product in an enzyme reaction. As a further example, some proteins are triggered by the binding of a substance known as an allosteric effector to adopt another conformational state that has substantially different activity (we explore protein function in Chapter 5). No matter how the folding is achieved, a particular amino acid sequence nearly always folds into the same conformation, and this conformation is required for proper protein function. Misfolding of proteins, whether due to mutations within the sequence or other factors, can affect protein function and could have serious consequences for the cell or organism.
Predicting Protein Folding Is a Goal of Computational Biology
The primary sequence holds the instructions for protein folding, so, theoretically, researchers should be able to predict a protein’s tertiary structure from its amino acid sequence alone. This is not yet possible, however—but not for want of trying. The protein folding “code” is complex, and the problem is complicated by the small difference in free energy between the folded and unfolded states of a protein. Although hydrogen bonds are strong, hydrogen bonding is not very different in the folded and unfolded states, because water is plentiful in the cell and can also hydrogen-bond with the protein. The main force driving protein folding is therefore derived from van der Waals contacts and the hydrophobic effect, but the free-energy difference between folded and unfolded protein states is still only about 5 to 15 kcal/mol. This small energy difference makes it difficult to understand and quantify the protein folding “code” that determines three-dimensional structure from primary sequence.
Inspection of many protein structures has provided some guidelines for protein folding. Hydrophobic residues avoid water by becoming buried, and this drives the polar polypeptide backbone into the interior, where it must form secondary structures with internal hydrogen bonds. The interior of a protein is amazingly well packed: about 75% of atoms in the interior of a protein are packed together as close as their van der Waals radii, the theoretical limit. Not even crystals of free amino acids have closer packing. Polar residues of a protein are usually at the surface, where the side chains can interact with water. These guidelines are used when predicting the extent to which different residues are buried in protein structures (Figure 4-19).
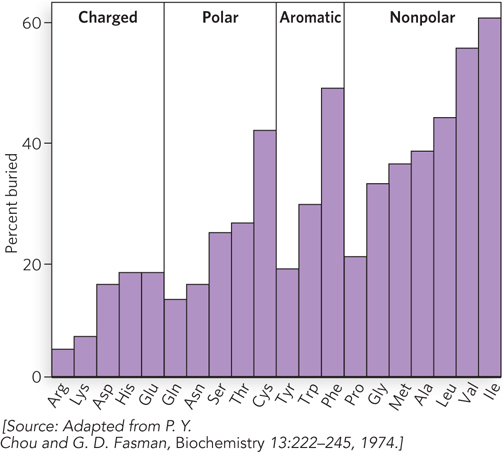
Figure 4-19: Buried residues. This plot shows the relative extent to which the 20 different amino acid residues are found in buried positions in globular proteins.
The exceptions to this general rule are easily explained. The hydrophobic side chain of proline is sometimes found on the surface, but Pro residues are useful for making sharp turns, which are usually on the surface. Cysteine is very moderately polar, but some Cys residues form disulfide bridges that are hydrophobic and therefore easily buried. Also, some residues that do not at first seem hydrophobic contribute greatly to hydrophobic forces. For example, lysine, a polar, charged amino acid, has a long hydrocarbon side chain that is often buried and contributes to hydrophobic interactions, while only its charged amino group protrudes to the surface.
Computational biologists have developed powerful algorithms to predict protein structure from amino acid sequences. The computations make use of the allowed values of torsion angles for different amino acids, their polar, ionic, and hydrophobic features, and the large body of structural information contained in the protein structure database. In fact, there is an international competition each year, called CASP (Critical Assessment of Protein Structure Prediction), which evaluates different protein structure prediction algorithms. To make this competition possible, research groups that have determined a protein structure but have not yet published it provide only the sequence of the protein to the CASP competition. Then computational biologists apply their algorithms to these sequences and arrive at structural models. When the experimentally determined structures are published, the degree to which the computed models agree with the experimentally determined structures is evaluated, and the scientist with the most accurate algorithm wins.
Besides CASP, there is a compelling protein-folding computational program set up as an online video game that enlists nonscientists in solving important questions in protein structure. The Foldit program for online gamers has even resulted in publication of original research papers in scientific journals. The Foldit puzzle game takes the beginner through a series of introductory puzzles that teach certain basic principles, then the user can choose from among several puzzles that enable nonscientists to help solve difficult computational science questions. This puzzle game is an experimental research project of the University of Washington Center for Game Science, in collaboration with the Biochemistry Department at the University of Washington. The premise is to add the human “insight” factor to computer-based simulation. At some point just prior to solution of a protein structure by x-ray crystallography, the protein sequence is presented to gamers. The object is to fold it correctly, and the proof is in the actual structure solved by a research lab. The algorithm has rules of folding from known structures, and the player can juggle, shake, and move sections of the protein around, scoring points as the protein backbone approaches a better fit to known parameters. Gamers typically form groups that chat and work together to achieve a high score within a set clock countdown. At the end of the competition, the highest score wins. But more exciting is when the actual structure is published, and gamers can see whether the high-scoring result actually arrived at the correct structure. In fact, winners of this game have also won first place in several categories of the CASP competition—all without the formal training of a biochemical or protein structural background.
The Foldit game also includes puzzles with experimental data and asks the gamer to fit the protein sequence into it. In 2011, Foldit players helped determine the crystal structure of an important drug target of a monkey AIDS virus, the retroviral protease of the Mason-Pfizer monkey virus. Technical hurdles prevented a solution of the crystal structure by conventional means, but the help of online gamers provided a solution that fit all known data in less than two weeks after the game was posted. This work led to a publication in a scientific journal.
Structure-based protein design is another goal of computational biology. The scientist starts with a “target” structure and then computationally designs a sequence that adopts the target folding pattern. Most of the algorithms developed for protein design focus on the redesign of a preexisting protein or “protein core,” the section that contains the central hydrophobic residues and defines how the protein folds. The complete de novo design of a protein is a difficult computational problem, given the vast number of possible conformations each amino acid residue can adopt.
A striking advance in de novo protein design was made by Steve Mayo’s group. An algorithm they had developed was applied to compute a sequence that folds into the structure of a zinc finger domain (Figure 4-20a). As we noted earlier in the chapter, zinc finger domains consist of a β-α-β motif, composed of about 30 amino acids. Despite its small size, the zinc finger domain contains sheet, helix, and turn structures, and the zinc atom is not required for proper folding. For a target structure, Mayo and colleagues used a 28-residue zinc finger domain in a protein called Zif268. There are 1.9 × 1027 possible combinations for 28-residue proteins, given the allowed torsion angles of the 20 common amino acids. The extent of the problem can be appreciated by the fact that just one molecule of each of these possible peptides, lumped together, would amount to 11.6 metric tons! The sequence to emerge from 90 hours of computing time gave a novel protein sequence that was unrelated to any known sequence in the database, yet was predicted to have a folding pattern similar to that of the target protein (compare the two patterns in Figure 4-20a, b). The protein having the computed sequence, named FSD1 (full sequence design 1), was then synthesized and the structure was experimentally determined by nuclear magnetic resonance (NMR) (Figure 4-20c); the actual FSD1 structure corresponded amazingly well with the target protein (Figure 4-20d). The overall deviation in backbone atoms of the target and FSD1 structures was only 0.98 Å over residues 8 through 26. The fact that completely different sequences can serve the same function supports the idea that once evolution arrives at a particular solution, it often uses this solution repeatedly in other proteins, rather than coming up with an entirely new one.
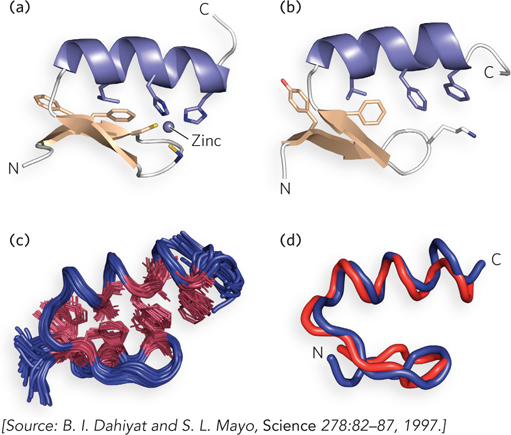
Figure 4-20: A protein structure designed by computation. (a) The structure of a zinc-binding domain from a DNA-binding protein (Zif268). (b) The predicted structure of the FSD1 sequence, derived entirely by computation. (c) The NMR structure of FSD1. (d) Superimposition of the Zif268 zinc-binding domain (red) and FSD1 (blue). Only residues 3 through 26 are shown.
Polypeptides Fold through a Molten Globule Intermediate
In 1968, Cyrus Levinthal reasoned that a protein should not be capable of randomly folding into a unique conformation in our lifetime. For example, starting from a 100-residue polypeptide in a random conformation, and assuming that each amino acid residue can have 10 different conformations, 10100 different conformations for the polypeptide are possible. If the protein folds randomly, by trying out all possible backbone conformations, and if each conformation is sampled in the shortest time possible (∼10−13 seconds, the time scale of a single molecular vibration), it would still take about 1077 years to sample all possible conformations! Yet, E. coli makes a biologically active protein of 100 amino acids in about 5 seconds at 37°C. This apparent contradiction is referred to as Levinthal’s paradox, and the astronomical disparity between calculation and observation reveals that protein folding is far from random: it must follow an ordered path that side-steps most of the possible intermediate conformations.
Intensive studies indicate the different ways that intermediate conformations could be side-stepped. We discuss here two pathways that are supported by experimental data. A hierarchical model of folding proposes that local regions of secondary structure form first, followed by longer-range interactions (e.g., between two α helices), and that this process continues until complete domains form and the entire polypeptide is folded (Figure 4-21).
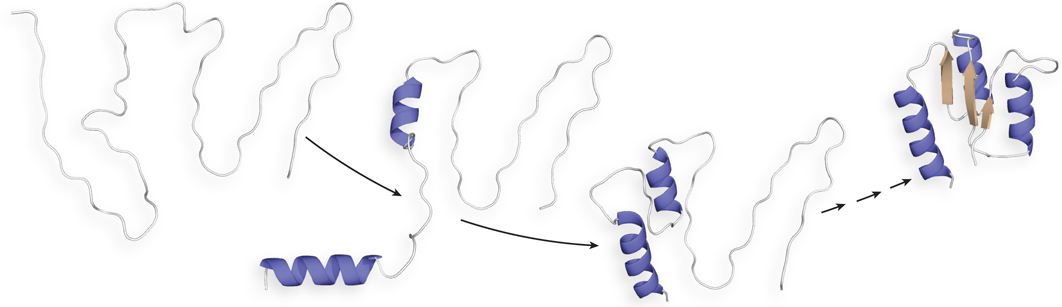
Figure 4-21: A hierarchical model of protein folding. In this model, which has been demonstrated for a few small proteins, some secondary structural elements form locally, and they nucleate the folding of the rest of the protein.
In the molten globule model, the hydrophobic residues of a polypeptide chain rapidly group together and collapse the chain into a condensed, partially ordered, “molten globule state.” With the protein condensed into a ball, the number of possible conformations is drastically limited to those that can occur within the confines of the molten globule. The molten globule state is not as compact as the final state; only about half of the hydrophobic residues are buried, and those that remain on the surface must find their way to the core. As hydrophobic residues are buried, the polar backbone atoms must form hydrogen-bonded secondary structures. The molten globule is therefore an ensemble of different partially ordered segments that shift and churn through multiple conformations, searching for the compact state of the native—completely folded—structure. As subdomains with tertiary structure begin to develop, the variety of different conformations decreases until most members of the population finally adopt the native structure.
Most proteins probably fold by a process that incorporates features of both models. Instead of following a single pathway, a population of identical polypeptides may take several routes to the same end point. Thermodynamically, the folding process can be viewed as a kind of free-energy funnel (Figure 4-22). Unfolded forms rapidly collapse to more compact forms, after which the number of conformational possibilities decreases. These rapid first stages quickly narrow the funnel. Small depressions along the sides of the free-energy funnel represent semistable intermediates that briefly slow the folding process. At the bottom of the funnel, the ensemble of folding intermediates has been reduced to the single conformation of the final, native state of the protein.
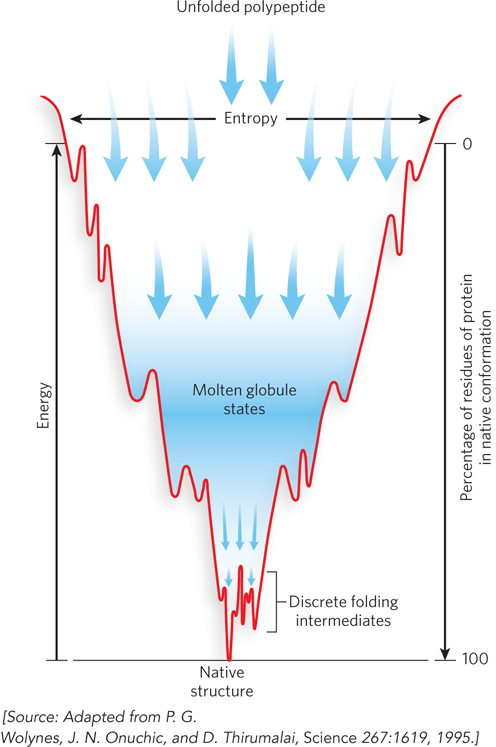
Figure 4-22: Protein folding as a free-energy funnel. The top of the funnel represents the unfolded state of a protein, the highest entropy state. As it folds, the protein progresses to lower free energy. The more compact the protein gets, the more rapidly it arrives at the properly folded and lowest free-energy state, at the bottom of the funnel.
Defects in protein folding may be the molecular basis for a wide range of human genetic disorders. For example, cystic fibrosis is caused by the misfolding of a chloride channel protein called cystic fibrosis transmembrane conductance regulator (CFTR). Many disease-related mutations in collagen are also caused by defective folding. In addition, defective protein folding causes the prion-related diseases of the brain (Highlight 4-3).
Thermodynamic stability is not evenly distributed over a protein. For example, a protein may have two stable domains joined by a segment with lower structural stability. The regions of low stability may allow the protein to alter its conformation between two (or more) states. As we shall see in Chapter 5, variations in the stability of regions within a given protein are often essential to protein function.
HIGHLIGHT 4-3 MEDICINE: Prion-Based Misfolding Diseases

Stanley Prusiner
A misfolded protein seems to be the cause of several rare, degenerative brain diseases in mammals. Perhaps the best known is bovine spongiform encephalopathy (BSE), also known as mad cow disease. An outbreak of BSE made international headlines in the spring of 1996. Related diseases are kuru and Creutzfeldt-Jakob disease in humans, scrapie in sheep, and chronic wasting disease in deer and elk. These diseases are referred to as spongiform encephalopathies, because the diseased brain frequently becomes riddled with holes and appears spongelike (Figure 1). Symptoms include dementia and loss of coordination, and the diseases are usually fatal.
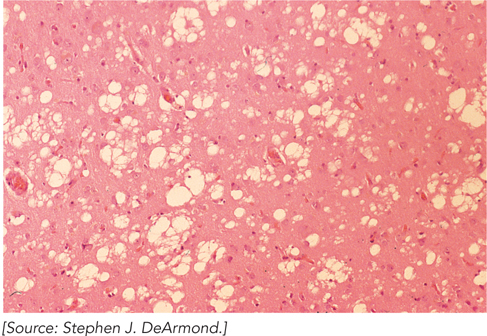
FIGURE 1 A stained section of cerebral cortex from a patient with Creutzfeldt-Jakob disease shows spongiform degeneration (holes in the tissue), the most characteristic neurohistological feature.
In the 1960s, investigators found that preparations of the disease-causing agents seemed to lack nucleic acids, suggesting that the agent was a protein. Initially, the idea seemed heretical. All disease-causing agents known up to that time—viruses, bacteria, fungi, and so on—contained nucleic acids, and their virulence was related to genetic reproduction and propagation. However, four decades of investigation, pursued most notably by Stanley Prusiner, provided evidence that spongiform encephalopathies are different.
The infectious agent has been traced to a single protein (Mr 28,000), dubbed prion (from proteinaceous infectious only; analogous to “virion”). The infected tissue contains abnormal spots with densely packed protein fibers called plaques. Fibers in these plaques are resistant to protease, and thus the protein is referred to as PrP (protease-resistant protein). The normal prion protein, called PrPC (C for cellular), is found throughout the body in healthy animals, and it does not form plaques. Its role in the mammalian brain (or any tissue) is not known, but it may have a molecular signaling function. Strains of mice lacking the Prnp gene that encodes PrP (and thus lacking the protein itself) suffer no obvious ill effects. Illness occurs only when the animal produces a PrP protein that misfolds, or converts, to an altered conformation, called PrPSc (Sc for scrapie). The misfolded PrPSc aggregates to form fibers that presumably lead to the plaques associated with spongiform encephalopathy. Although researchers do not completely understand how the misfolded state of PrP is propagated during infection, it is commonly thought that the interaction of PrPSc with PrPC converts the latter to PrPSc, initiating a domino effect in which more and more of the brain protein converts to the disease-causing form.
The structure of the C-terminal region of normal PrPC is known (Figure 2); it contains three α helices and two β strands. In contrast, fibers of PrPSc, as shown by circular dichroism, contain a core of tightly packed β sheets. The exact structure of PrPSc is unknown because it is an aggregate and difficult to work with, but it is thought to contain several β sheets—accounting for the prevalence of β sheet structure in the PrPSc aggregate inferred from CD measurements. Future insights about the structure and function of PrPSc will provide treatment strategies for this disease.
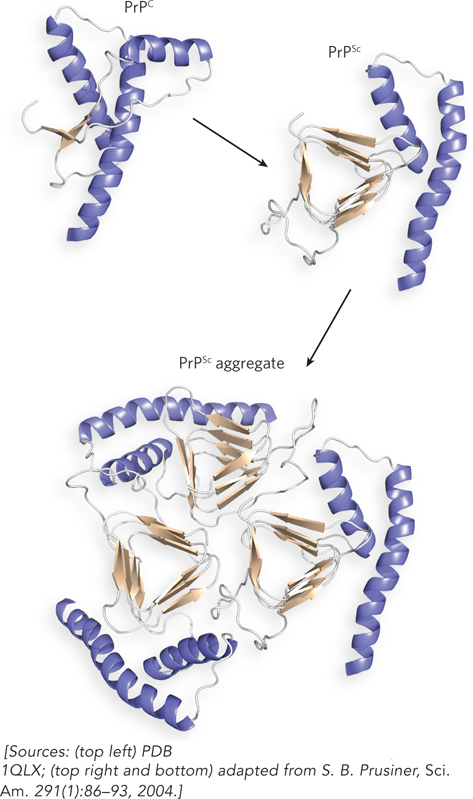
FIGURE 2 The globular domain of human PrPC monomer (top left) contains three α helices and two short β strands. To its right is a proposed structure of the corresponding region of PrPSc, containing many more β strands, which may promote aggregation (bottom).
Spongiform encephalopathy can be inherited, can occur spontaneously, or can be transmitted through infection. Most cases are spontaneous. Infectious transmission accounts for fewer than 1% of cases and occurs through direct contact with diseased tissue. Inherited forms of prion diseases, which account for 10% to 15% of cases, are due to a variety of point mutations in the Prnp gene, each of which is believed to make the spontaneous conversion of PrPC to PrPSc more likely. A detailed understanding of prion diseases awaits new information on how prions affect brain function. One recent advance is the development of mouse models of the disease. With the alteration of just one amino acid residue, the mutant prion protein leads to disease that faithfully mimics the full disease spectrum, from formation of plaques in the brain to transmission of disease to healthy animals by direct contact. Mouse models hold promise for developing and testing therapies to prevent these fatal neurodegenerative diseases.
Chaperones and Chaperonins Can Facilitate Protein Folding
Not all proteins fold spontaneously. The folding of many proteins is facilitated by chaperones, specialized proteins that bind improperly folded polypeptides and facilitate correct folding pathways or provide microenvironments in which folding can occur. Two classes of molecular chaperones have been well studied, and both are conserved across species, from bacteria to humans. Proteins in the first class, a family of proteins called Hsp70 (heat shock proteins of Mr 70,000), become more abundant in cells stressed by elevated temperature. Hsp70 proteins bind to unfolded regions that are rich in hydrophobic residues, preventing aggregation and thus protecting denatured proteins and newly forming proteins that are not yet folded. Some chaperones also facilitate the assembly of subunits in oligomeric proteins.
Hsp70 proteins bind and release polypeptides in a cycle that involves several other proteins (including the class Hsp40) and ATP hydrolysis. Figure 4-23 diagrams chaperone-assisted folding of a protein mediated by the DnaK and DnaJ chaperones of E. coli, functional equivalents (homologs) of the eukaryotic Hsp70 and Hsp40 proteins.
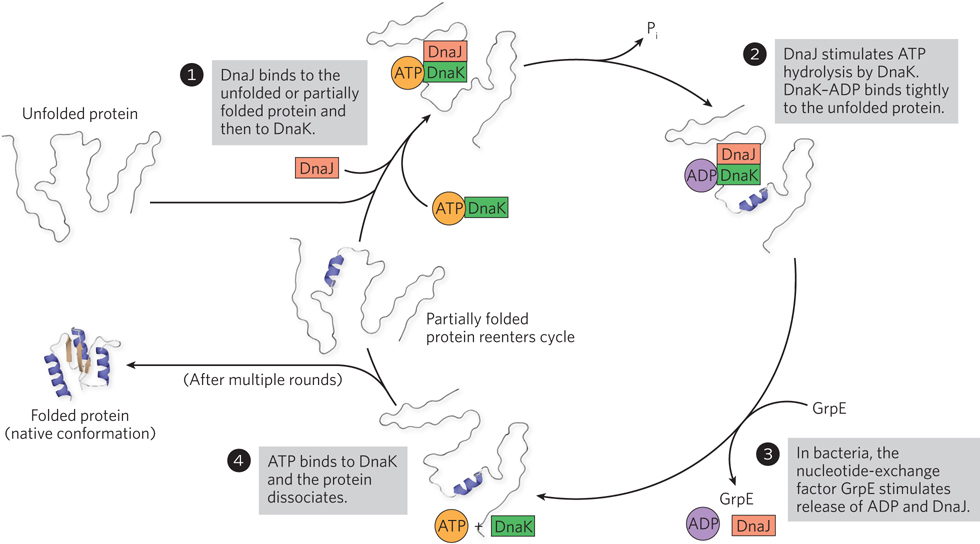
Figure 4-23: Chaperone-assisted protein folding. Chaperones bind and release polypeptides by a cyclic pathway, shown here for E. coli chaperone proteins DnaK and DnaJ, homologs of the eukaryotic chaperones Hsp70 and Hsp40. The chaperones mainly prevent the aggregation of unfolded polypeptides. Some polypeptides released at the end of a cycle are in their native conformation; the rest are rebound by DnaJ and DnaK to repeat the cycle for further attempts at refolding. In bacteria, GrpE interacts transiently with DnaK late in the cycle (step 3), promoting dissociation of ADP and possibly DnaJ. No eukaryotic analog of GrpE is known.
Chaperonins, the second class of chaperones, are elaborate protein complexes required for the folding of some cellular proteins. In E. coli, an estimated 10% to 15% of cellular proteins require the resident chaperonin system—GroEL/GroES—for folding under normal conditions. Up to 30% of proteins require assistance when the cell is heat-stressed. Unfolded proteins are bound in pockets in the GroEL complex, and the pockets are capped transiently by GroES (Figure 4-24). GroEL undergoes substantial conformational changes, coupled with ATP hydrolysis and the binding and release of GroES, which together promote folding of the bound polypeptide. Although the structure of the GroEL/GroES chaperonin is known, many details of its mechanism of action remain unresolved.
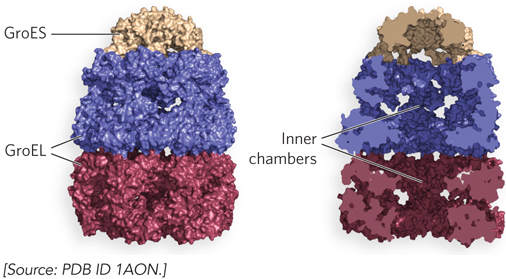
Figure 4-24: Chaperonin-assisted protein folding. The E. coli chaperonins GroEL and GroES. Each GroEL complex consists of two chambers formed by two heptameric rings (each subunit Mr 57,000). GroES, also a heptamer (each subunit Mr 10,000), blocks one of the GroEL pockets. Surface (left) and cut-away (right) images of the GroEL/GroES complex are shown.
Protein Isomerases Assist in the Folding of Some Proteins
The folding of certain proteins requires enzymes that catalyze isomerization reactions. Protein disulfide isomerase is a widely distributed enzyme that catalyzes the interchange, or shuffling, of disulfide bonds until the bonds of a protein’s final conformation have formed. Among its functions, protein disulfide isomerase catalyzes the elimination of folding intermediates with inappropriate disulfide cross-links.
The cis and trans isomers of peptide bonds are rapidly interchangeable (see Section 4.1). However, proline’s cyclic structure makes interconversion between cis and trans isomers a slow process. Peptide prolyl cis-trans isomerase catalyzes the interconversion of the cis and trans isomers of proline peptide bonds (Figure 4-25). Most peptide bonds in proteins are in the trans isomeric form, but the cis isomer of proline is often found in tight turns between secondary structural elements.
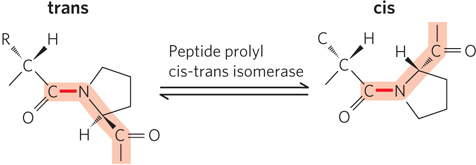
Figure 4-25: The trans and cis isomers of proline in a peptide bond. Most peptide bonds (>99.95%) are in the trans configuration, but about 6% of bonds involving Pro residues are in the cis configuration; many of these occur in β turns.
SECTION 4.4 SUMMARY
The polypeptide chain of a protein folds into a unique conformation, and the instructions for this folding are inherent in its primary sequence.
The folding pattern of a protein is hard to predict from the primary sequence, because the forces that stabilize the folded state are weak and cannot be recognized from the amino acid sequence.
In a protein’s folded state, hydrophobic residues are usually found in the interior of the protein, and polar residues often localize to the surface.
The protein-folding pathway is not random. The folding of some proteins is thought to proceed through a condensed molten globule state that limits folding to conformations that are compatible with a compact volume.
Protein folding is sometimes assisted by chaperones and chaperonins. Chaperones are proteins of the Hsp70 class that bind unfolded proteins and use cycles of ATP-binding and hydrolysis to help the proteins refold. Chaperonins are complex multisubunit structures that engulf the protein in a chamber during the refolding process.
Protein folding is also assisted by isomerases. Protein disulfide isomerase catalyzes the breakage and reformation of disulfide bonds, and peptide prolyl cis-trans isomerase facilitates interchange between the cis and trans isomers of Pro residues.