5.4 The Regulation of Protein Function
In the flow of biological information, groups of enzymes often work together in sequential and interconnected pathways to carry out a given process, such as the replication of a chromosome or the splicing of an intron in a messenger RNA. Such processes use enormous amounts of chemical energy in the form of nucleoside triphosphates (NTPs). It is critical not only that these processes occur, but that they do so only at a specific time and in a specific place, so that resources are not wasted. In addition, the many reactions that carry out these complex processes must be precisely coordinated. Faulty coordination or poor timing could damage or alter the cellular genome. Regulation is thus an important aspect of virtually every process in molecular biology.
158
Most enzymes follow the Michaelis-
The activities of many proteins in DNA and RNA metabolism are regulated, including binding proteins that simply bind reversibly to DNA or RNA, enzymes, and motor proteins. Their functions can be modulated in several ways: by the noncovalent binding of allosteric modulators, autoinhibition by a segment of the protein, reversible covalent modification, proteolytic cleavage, or interaction with special regulatory proteins. We touch on all of these mechanisms, while focusing on those that are most common in enzymes and proteins involved in nucleic acid metabolism.
Modulator Binding Causes Conformational Changes in Allosteric Proteins
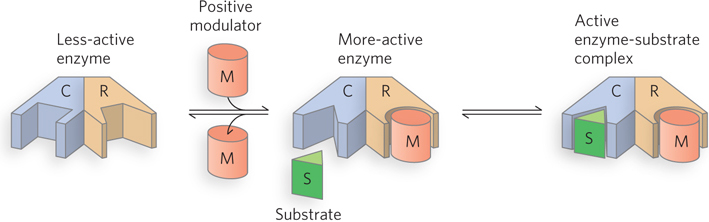
Allosteric enzymes or allosteric proteins are those having “other shapes” or other conformations induced by the binding of modulators. This property is found in certain regulatory enzymes, as conformational changes induced by one or more allosteric modulators interconvert more-
The properties of allosteric enzymes are significantly different from those of simple, nonregulatory enzymes. Some of the differences are structural. In addition to active sites, allosteric enzymes generally have one or more regulatory, or allosteric, sites for binding the modulator (Figure 5-18). Just as an enzyme’s active site is specific for its substrate, each regulatory site is specific for its modulator. Enzymes with several modulators generally have different specific binding sites for each.
Allosteric Enzymes Have Distinctive Binding and/or Kinetic Properties
Allosteric enzymes show relationships between V0 and [S] that differ from Michaelis-
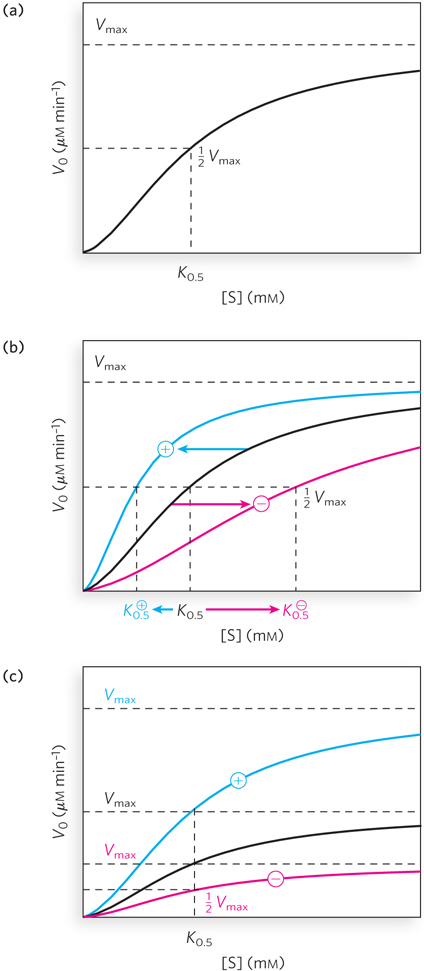
For homotropic allosteric proteins or enzymes, the substrate often acts as a positive modulator (an activator), because the subunits act cooperatively. Cooperativity occurs when the binding of a substrate to one binding site alters an enzyme’s conformation and affects the binding of subsequent substrate molecules. Most commonly, the binding of one molecule enhances the binding of others, an effect called positive cooperativity. This accounts for the sigmoid rather than hyperbolic change in V0 with increasing [S]. One characteristic of sigmoid kinetics is that small changes in the concentration of a modulator can be associated with large changes in enzyme activity. A relatively small increase in [S] in the steep part of the curve causes a comparatively large increase in V0 (see Figure 5-19a). Much rarer are cases of negative cooperativity, where the binding of one substrate molecule impedes the binding of subsequent molecules.
159
For heterotropic allosteric proteins or enzymes, those with modulators that are molecules other than the normal substrate, it is difficult to generalize about the shape of the binding or substrate-
Many of the allosteric effects encountered by molecular biologists are heterotropic. Numerous examples can be found among the regulatory proteins that bind to specific DNA sequences adjacent to genes, such as the cAMP receptor protein, or CRP (also called CAP, for catabolite gene activation protein; see Figure 4-18a). CRP is a dimeric DNA-
Autoinhibition Can Affect Enzyme Activity
Many processes of DNA and RNA metabolism are precisely targeted; they are limited to particular locations and circumstances, some of which appear transiently and unpredictably. For example, if DNA is damaged, the lesion must be repaired before the next replication cycle. The enzymes that repair DNA cleave the DNA strands near the lesion, remove damaged nucleotides, and replace them. It is essential that these enzymes be available on short notice and that they act only at DNA lesions.
160
One way to make such enzymes generally available but not active until needed is by autoinhibition. A segment of the protein molecule, sometimes an entire domain, can reduce or eliminate the activity of the enzyme. The enzyme may be present in the cell at all times, but it functions weakly or not at all under normal circumstances. Activation of the enzyme requires self-
Autoinhibition has been documented for several proteins, including certain helicases and the bacterial RecA recombinase. In bacteria, the activity of a helicase known as Rep, involved in DNA replication, is autoinhibited by a subdomain called 2B. The bacterial RecA recombinase is autoinhibited by a short segment of polypeptide at its C-
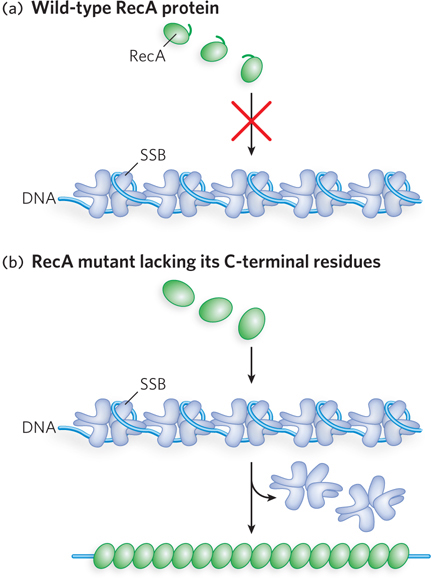
Autoinhibition may be just one aspect of a broader regulatory strategy. The autoinhibited enzyme can be maintained in the cell without its activity causing problems when it is not needed. Activation by interaction with additional proteins can occur quickly when its activity is required, and the enzyme’s function can be more readily targeted to specified locations and situations.
Some Proteins Are Regulated by Reversible Covalent Modification
In another important class of regulatory mechanism, activity is modulated by covalent modification of one or more amino acid residues in a protein molecule. More than 300 different types of covalent modification have been found in proteins. Common modifying groups include phosphoryl, acetyl, adenylyl, uridylyl, methyl, amide, carboxyl, myristoyl, palmitoyl, prenyl, hydroxyl, sulfate, and adenosine diphosphate ribosyl groups. There are even entire proteins that are used as specialized modifying groups, such as ubiquitin. Some of these modifications are shown in Figure 5-21. These varied groups are generally linked to and removed from a regulated enzyme or other protein by separate enzymes. When an amino acid residue is modified, a novel amino acid with altered properties is effectively introduced into the protein. The introduction of a charge can alter an enzyme’s local properties and induce a conformational change. The introduction of a hydrophobic group can trigger association with a membrane. The changes are often substantial and can be critical to the function of the altered enzyme.
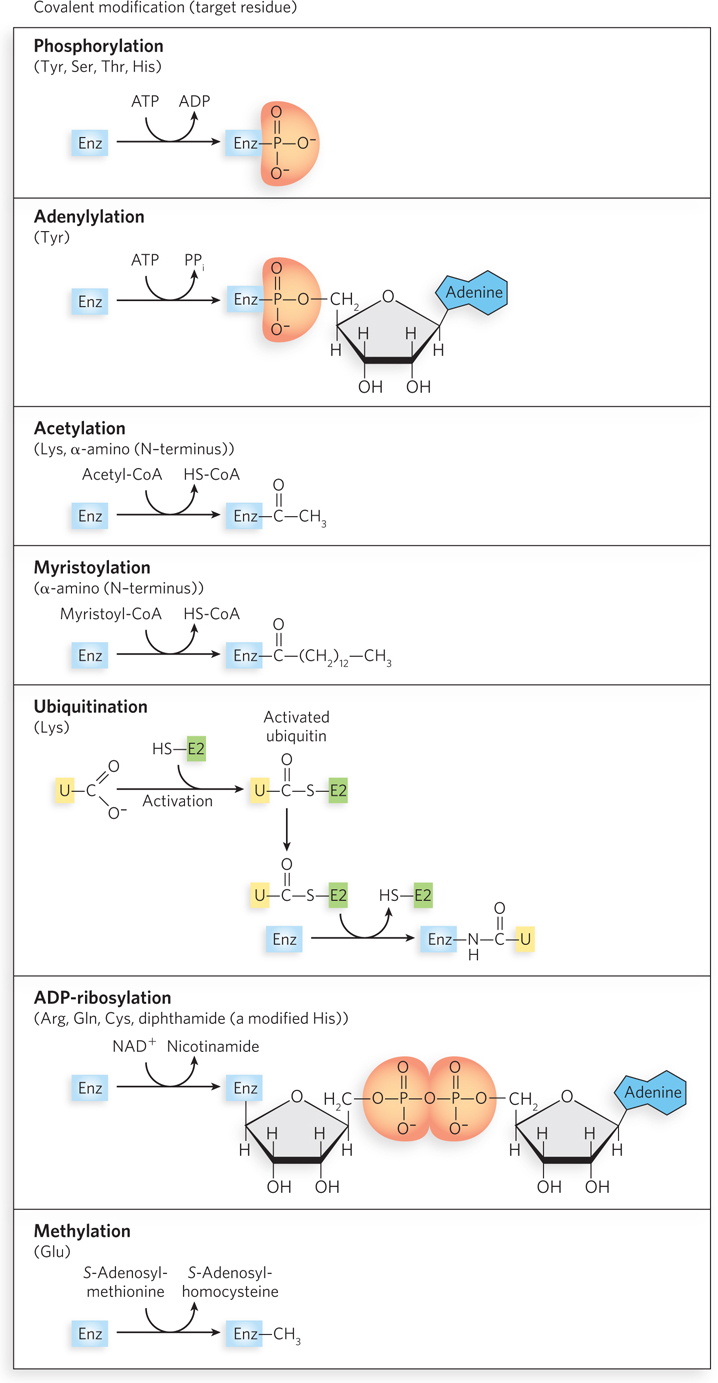
The variety of protein modifications is too great to cover in detail, but we present a few examples here. In eukaryotic cells, histones are important modification targets. As described in detail in Chapter 10, many histones and histone variants are subject to precise patterns of methylation, acetylation, phosphorylation, and ubiquitination. Such modifications play an important role in altering chromatin structure in specific regions, to facilitate gene expression and other activities.
161
162
Phosphorylation is probably the most common type of regulatory modification. It is estimated that one-
Phosphoryl Groups Affect the Structure and Catalytic Activity of Proteins
The attachment of phosphoryl groups to specific amino acid residues of a protein is catalyzed by protein kinases; the removal of the groups is catalyzed by protein phosphatases. The addition of a phosphoryl group to a Ser, Thr, or Tyr residue introduces a bulky, charged group into a region that was previously only moderately polar. The oxygen atoms of a phosphoryl group can hydrogen-
The Ser, Thr, or Tyr residues that are phosphorylated in regulated proteins occur within common structural motifs called consensus sequences that are recognized by specific protein kinases. Some kinases are basophilic, preferentially phosphorylating a residue that has basic neighbors; others have different substrate preferences, such as for a residue near a proline. Besides local amino acid sequence, the overall three-
To serve as an effective regulatory mechanism, phosphorylation must be reversible. Cells contain a family of phosphoprotein phosphatases that hydrolyze specific  –Ser,
Thr, and
Tyr esters (
is shorthand for the phosphoryl group), releasing inorganic phosphate (Pi). The phosphoprotein phosphatases we know of thus far act only on a subset of phosphoproteins, but they show less substrate specificity than protein kinases.
–Ser,
Thr, and
Tyr esters (
is shorthand for the phosphoryl group), releasing inorganic phosphate (Pi). The phosphoprotein phosphatases we know of thus far act only on a subset of phosphoproteins, but they show less substrate specificity than protein kinases.
Some Proteins Are Regulated by Proteolytic Cleavage
In the process of proteolytic cleavage, an inactive precursor protein is cleaved to form the active protein. Many eukaryotic proteases (proteolytic enzymes) are regulated in this way. A subunit of the E. coli DNA polymerase V (see Chapter 12) called MutD is also activated in this way, with cleavage producing the active form, MutD′.
The larger, uncleaved precursor proteins, before proteolytic cleavage, are generally referred to as proproteins or proenzymes, as appropriate. For example, a class of proteins known as transcription factors facilitate the function of RNA polymerases in all organisms. The process of sporulation (spore formation) in the bacterium Bacillus subtilis is controlled in part by a transcription factor called σE, which is synthesized as an inactive proprotein, pro-
As another example, the small number of proteins that are encoded by the genomes of eukaryotic retroviruses, including the human immunodeficiency virus, HIV, are generally synthesized as one large polyprotein, which must be cleaved into the individual functional proteins by a virus-
HIGHLIGHT 5-2 MEDICINE: HIV Protease: Rational Drug Design Using Protein Structure
Human immunodeficiency virus (HIV), the causative agent of AIDS, kills cells of the immune system. The development of a vaccine has been unsuccessful, because the surface glycoproteins targeted by antibodies change rapidly, in part due to the extremely high mutation rate of HIV (about one replication mistake in every 10,000 nucleotides of HIV genome per generation). However, there has been substantial success in the development of drugs targeting HIV-
The usual route for developing a drug that inhibits enzyme activity starts with the random screening of hundreds of thousands of chemical compounds. Possible inhibitors are then chemically optimized for potency, availability in an oral form, and low toxicity. The U.S. Food and Drug Administration’s approval procedure for the use of a drug in humans usually takes well over a dozen years. This type of process has led to drugs that inhibit certain HIV enzymes, including the reverse transcriptase. Rational drug design is another, shorter route to drug discovery. It starts with the target protein structure and designs chemicals that plug the active site and shut the enzyme down. This process short-
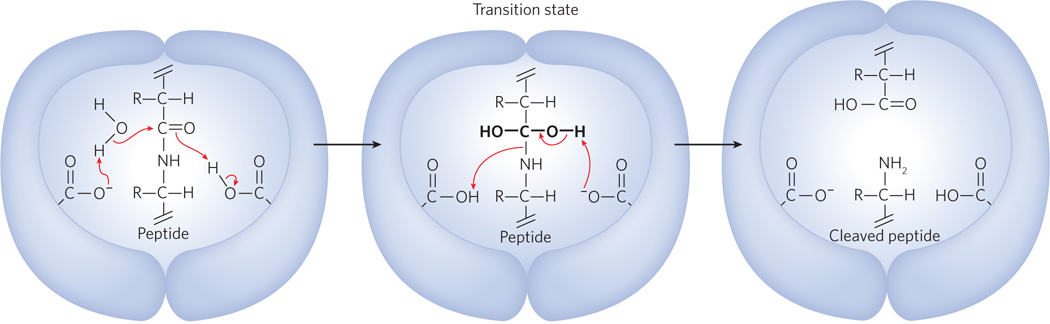
An astounding success in rational drug design has been achieved with the HIV protease. This is partly due to the unique architecture of the protein. HIV protease is a dimer of identical subunits, but unlike typical dimers, which contain two active sites, the HIV protease dimer has a single active site located in the central hydrophobic chamber at the dimer interface. The active site has twofold symmetry; each subunit contributes a catalytic Asp residue, and the two cooperate to hydrolyze the peptide bond (Figure 1). Information obtained from the biochemical studies and crystal structures of HIV protease made possible the rational design of chemical inhibitors. By 1996, the FDA had approved three HIV protease inhibitor drugs: indinavir (Crixivan), ritonavir (Norvir), and saquinavir (Invirase). Notably, all of these drugs are effective, in part, because they mimic the transition state of the proteolytic reaction catalyzed by the enzyme and thus bind to the enzyme virtually irreversibly (Figure 2).
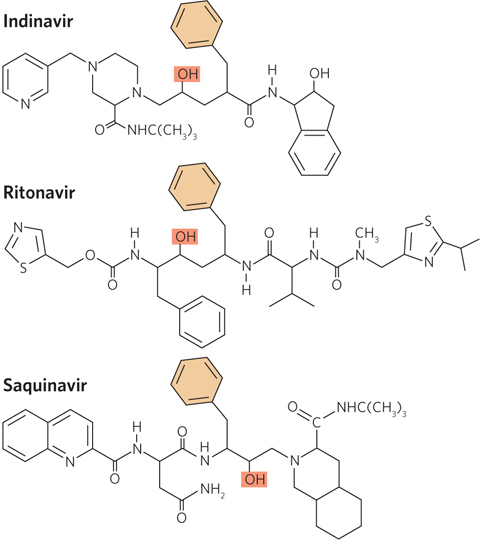
Protease inhibitor drugs have helped reduce the viral titer in the plasma of HIV-
SECTION 5.4 SUMMARY
Specific enzymes, motor proteins, and other proteins are subject to various types of regulation.
The noncovalent binding of allosteric modulators, either homotropic or heterotropic, can facilitate or inhibit the activity of nucleic acid–
binding proteins or enzymes. Parts of a protein’s own structure can reduce the overall activity of the protein in the process of autoinhibition.
Covalent modification is a common mechanism used to alter the function of proteins and enzymes. Common modifications involve the addition and removal of phosphoryl, methyl, acetyl, ubiquitinyl, and many other types of groups.
Ser, Thr, and Tyr residues can be phosphorylated and dephosphorylated by protein kinases and phosphatases, respectively. Kinases are specific to consensus sequences in the target protein, but phosphorylases are less specific.
Some proteins and enzymes are regulated by proteolytic cleavage. These proteins are synthesized as larger, inactive proproteins or proenzymes and are activated by the proteolytic removal of one or more amino acid residues.
163
UNANSWERED QUESTIONS
The study of protein function is, arguably, the oldest subdiscipline in biochemistry and molecular biology. But there is still much to learn. The relatively young science of genomics keeps pointing to genes that encode proteins about which we know little or nothing at all. Some shortcuts to functional discovery are discussed in later chapters.
How does protein structure relate to function? This is an old but still very relevant question for every scientist who studies proteins. Advanced methods of structural analysis are providing more information than ever before, but many of these structural pictures are static. A clear picture of a complete binding or catalytic cycle can require a detailed knowledge of the structure of multiple protein conformations. Certain structural motifs and domains (e.g., the OB fold of single-
stranded DNA– binding proteins and other proteins, the AAA+ ATPase domain, and simple β-barrel structures) appear in proteins that often have seemingly unrelated functions. The manner in which particular structures are adapted to different functions is an ongoing area of investigation. How do proteins function in the context of large protein assemblies? Many proteins act only as a part of a much larger protein complex, involving anywhere from a few to many dozens of additional proteins. Unraveling the individual contributions of the subunits of these large complexes has become one of the major challenges of modern molecular biology.
Within the context of molecular biology, how many types of protein function remain to be discovered? A textbook such as this one might leave a student with the impression that the fundamental protein/enzyme activities underlying information pathways are now understood. This is not so! Although major processes such as DNA replication, RNA transcription, and protein synthesis are increasingly well understood, new types of proteins with important functions are continually being discovered. Many of the newer discoveries involve proteins that have regulatory functions, or facilitate changes in the bacterial nucleoid or eukaryotic chromatin during cell division, or carry out functions in RNA metabolism. There are no boundaries to these frontiers of protein research.
How do proteins in the small concentrations found in cells find their interacting partners—
particularly, specific sequences on very large nucleic acids— in the complex cellular environment? This is an issue that remains of great interest to investigators in many areas of molecular biology.
164
165
166
The Discovery of the Lactose Repressor: One of the Great Sagas of Molecular Biology
Rickenberg, H.V., G.N. Cohen, G. Buttin, and J. Monod. 1956. La galactoside-

Jacques Monod began his scientific career in the 1930s, as a graduate student with André Lwoff, studying the capacity of E. coli to adapt its metabolism to different growth conditions. His approach to science was shaped in part by a 1936 trip to Thomas Hunt Morgan’s laboratory at the California Institute of Technology, where he found a stimulating environment dominated by open collaboration and free discussion. Back in France, Monod’s scientific career was slowed but not halted by the outbreak of World War II. While continuing his research, Monod was an active member of the French Resistance. His laboratory at the Sorbonne doubled as a meeting place and propaganda printing press. In the lab, he hit on the idea of an inducer, a cellular signal that would trigger the production of new enzymes needed for adapting to new metabolic circumstances. After the war, he turned back to science full-
The disaccharide lactose is cleaved to the monosaccharides glucose and galactose by the enzyme β-galactosidase. Monod found that when lactose was not present in the E. coli growth medium, β-galactosidase was barely detectable in cell extracts. When lactose was added as the sole carbon source for bacterial growth, the levels of β-galactosidase enzyme increased dramatically. Monod wondered how this might occur. In a 1940s’ scientific world in which DNA sequencing, PCR (the polymerase chain reaction), and the structure of DNA were unknown, and messenger RNA still remained to be discovered, the question was not trivial.
Many bacterial enzymes of intermediary metabolism exhibit this inducible pattern. What recommended lactose metabolism as a subject of investigation? Like many other laboratories in the late 1940s, the Pasteur Institute lacked modern cold rooms and other facilities now commonly used to keep proteins active during purification. The one inducible enzyme stable enough to survive the summer heat of an attic lab in Paris was β-galactosidase, later shown to be encoded by a gene called lacZ. The enzyme was also fairly easy to assay. When it was present at high levels, it would cleave an alternative substrate, 5-
Mutations were soon found that affected the induction of β-galactosidase. Colonies of these mutants turned blue on X-
167
The lacI Gene Encodes a Repressor
Jacob, F., and J. Monod. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3:318–
Next door to Monod at the Pasteur Institute was the laboratory of François Jacob. Wounded as a member of the Free French Forces during the Normandy campaign, Jacob could not pursue his planned career in surgery and instead turned to science. He studied a phenomenon dubbed the “erotic induction” of bacteriophage λ. Bacteriophage λ is a prophage, a bacterial virus that integrates its DNA into its host chromosome and remains quiescent there.
Some years earlier, Joshua Lederberg had discovered the phenomenon of bacterial conjugation (bacterial sex), in which DNA is transferred from a donor to a recipient cell. The transfer is mediated by a genetic element, separate from the bacterial chromosome, known as the F plasmid. In some cells, this plasmid becomes integrated into the host chromosome, creating a strain that mediates high-
Jacob, working with Elie Wollman, used this technique to produce some of the first genetic maps, and they even deduced that the E. coli chromosome was circular. They also noticed that when an Hfr strain also contained an integrated prophage, the bacteriophage λ was transferred into the recipient along with the chromosomal genes. When the dormant bacteriophage λ was thus transferred to a strain that lacked an integrated prophage, the recipient cells were soon lysed. The prophage was somehow activated when it entered the recipient, leading to lytic reproduction—
Monod first liked the idea that mutation in the lacI gene (lacI−) produced some kind of inducer that made the addition of lactose unnecessary. If this was true, then the lacI− gene should be dominant over the normal (wild-
The work continued. If lacI was encoding a repressor, this repressor had to interact with something to shut down the lactose genes. Monod and Jacob now mutagenized cells that had two good copies of the lactose genes. It was unlikely that both copies of lacI would be inactivated, but inactivation of one repressor target would be enough to induce the function of one set of lactose genes. The researchers predicted that the resulting mutations would appear at a site on the chromosome distinct from lacI and would lead to constitutive synthesis of the lactose gene products. The mutants were found, and they defined a site that Monod and Jacob called the operator.
In a famous 1961 paper, Jacob and Monod laid out these ideas and others as part of their operon model. The concepts have guided our thinking about gene regulation ever since. Other experiments, carried out in parallel, showed that bacteriophage λ also encodes a repressor, and this repressor is needed to keep most other bacteriophage λ genes from being expressed.
168
Discovery of the Lactose Repressor Helped Give Rise to DNA Sequencing
Gilbert, W., and B. Müller-

By 1961, it was clear that a repressor existed, but not at all clear what it was. It could be RNA or protein, or some other kind of molecule that was synthesized by a lacIenzyme. By 1966, nonsense-
To isolate a protein, you need a way to measure its presence (an assay), but that was difficult to construct. The repressor presumably bound to operator DNA, but that DNA sequence was not yet defined. The repressor also bound to the inducer (then thought to be lactose, later shown to be allolactose, a metabolic by-
Using a technique known as equilibrium dialysis, the researchers suspended a dialysis bag containing a bacterial cell extract in a solution containing radioactive IPTG. Pores in the dialysis bag were sufficiently small to prevent the protein molecules from escaping, but smaller molecules such as IPTG could diffuse through. If the lactose repressor was present in the extract, it would bind to IPTG, and the concentration of IPTG would increase inside the dialysis bag relative to the surrounding solution. The assay eventually worked, but only after Gilbert and his colleagues developed methods to greatly increase its sensitivity. In 1966, they reported their detection of a repressor protein that bound IPTG.
The lactose repressor was finally purified to homogeneity by the Gilbert group and several other research groups in the early 1970s. Gilbert used the repressor problem to develop several new methods to define the DNA binding sites of proteins that bound DNA specifically. For example, his group found that when dimethylsulfate (DMS) was used to modify adenine or guanine residues in the DNA, the adjacent DNA backbone became more labile to cleavage in mild alkali. DMS methylates guanine residues at N-
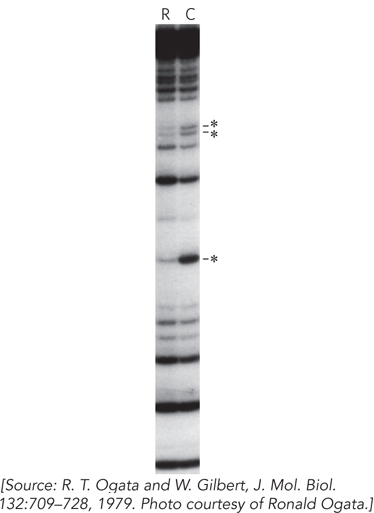
In this technique, the control lane (lane C in Figure 2) proved to be as important as the protein that was the subject of the experiment. As Gilbert and his colleague Allan Maxam looked at one of these gels, they realized that the gel could be read to reveal the positions of all the A and G residues in the DNA strand. If they could find a technique to break the strands at C and T residues as well, they would have a new way to sequence DNA. The methods were developed and published as a new sequencing technology in 1977. The Maxam-
169