7.1 ISOLATING GENES FOR STUDY (CLONING)
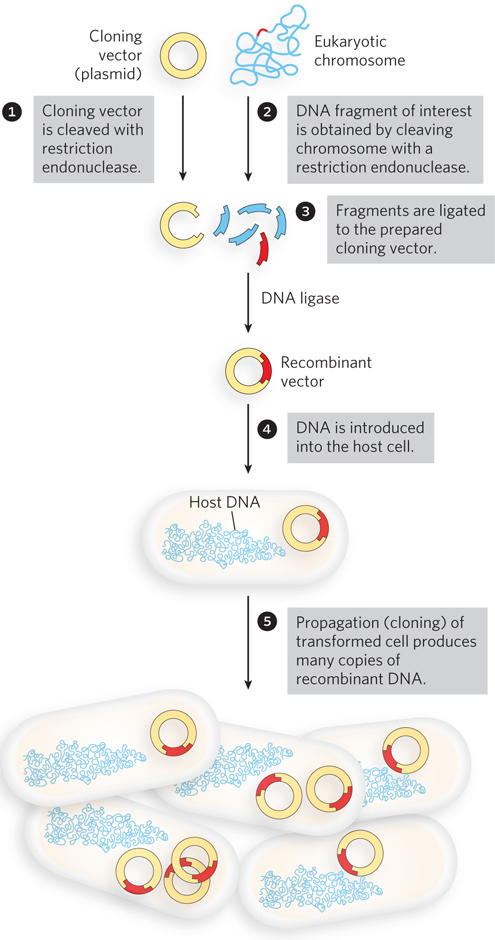
A clone is an identical copy. The term was originally applied to cells produced when a cell of a single type was isolated and allowed to reproduce to create a population of identical cells. DNA cloning involves separating a specific gene or DNA segment from a larger chromosome, attaching it to a small molecule of carrier DNA, introducing this modified DNA into a host cell, then replicating the DNA by increasing both the cell number and the copy number of the cloned DNA in each cell. The result is selective amplification of a particular gene or DNA segment.
213
The cloning of DNA from any organism entails five general steps:
Obtaining the DNA segment to be cloned. Enzymes called restriction endonucleases act as precise molecular scissors, recognizing specific sequences in DNA and cleaving genomic DNA into smaller fragments suitable for cloning. Alternatively, genomic DNA may be sheared randomly into fragments of a desired size. Or, since the sequence of targeted genomic regions is often known, some DNA segments to be cloned are simply synthesized.
Selecting a small molecule of DNA capable of self-
replication. These small DNAs are called cloning vectors (a vector is a carrier or delivery agent). Most cloning vectors used in the laboratory are modified versions of naturally occurring small DNA molecules found in bacteria and lower eukaryotes such as yeast. Small viral DNAs may also play this role.Joining two DNA fragments covalently. The enzyme DNA ligase links the cloning vector to the DNA fragment to be cloned. Composite DNA molecules of this type, comprising covalently linked segments from two or more sources, are called recombinant DNAs.
Moving recombinant DNA from the test tube to a host organism. The host organism provides the enzymatic machinery for DNA replication.
Selecting or identifying host cells that contain recombinant DNA. The cloning vector generally has features that allow the host cells to survive in an environment where cells lacking the vector would die. Cells containing the vector are thus “selectable” in that environment.
The methods used for accomplishing these and related tasks are collectively referred to as recombinant DNA technology or, more informally, genetic engineering.
Much of our initial discussion focuses on DNA cloning in the bacterium Escherichia coli, the first organism used for recombinant DNA work and still the most common host cell. E. coli has many advantages. Its DNA metabolism (like many of its other biochemical processes) is well understood, many naturally occurring cloning vectors associated with this bacterium are well characterized, and techniques are available for easily moving DNA from one bacterial cell to another. The principles discussed here are also broadly applicable to DNA cloning in other organisms, as we will see later in the chapter.
Genes Are Cloned by Insertion into Cloning Vectors
DNA can be cloned from any cellular or viral source. Although the approaches are determined partly by the DNA source and what is known about it, all cloning efforts have a few enzymes and procedures in common. Recombinant DNA technology relies on a set of enzymes made available through decades of research on nucleic acid metabolism (Table 7-1). Two classes of enzymes are particularly important: the restriction endonucleases (restriction enzymes) and DNA ligase (Figure 7-1). First, restriction endonucleases recognize DNA at specific recognition sequences (or restriction sites) and cleave it to generate a set of smaller fragments. Second, a DNA fragment of interest can be joined to the DNA of a suitable cloning vector by DNA ligase. The recombinant vector is then introduced into a host cell, which amplifies the DNA fragment in the course of many generations of cell division.

214
Restriction endonucleases are found in a wide range of bacterial species. Werner Arber discovered in the early 1960s that the biological function of these enzymes is to recognize and cleave foreign DNA (the DNA of an infecting virus, for example); such DNA is said to be restricted. Acting in a system with other enzymes that protect the host DNA, restriction endonucleases participate in a kind of immune system in bacteria. There are three types of restriction endonucleases, distinguished by their complexity and the typical distance between recognition sequence and cleavage site. Type II restriction endonucleases, first reported by Hamilton Smith in 1970, are the simplest, require no ATP for their activity, and cleave the DNA within the recognition sequence. Daniel Nathans quickly put this group of restriction endonucleases to use, demonstrating their extraordinary utility by developing novel methods for mapping and analyzing genes and genomes.
Thousands of restriction endonucleases have been discovered in different bacterial species, and more than 100 different DNA sequences are recognized by one or more of these enzymes. The recognition sequences are usually 4 to 8 base pairs (bp) long and palindromic (the recognition sequence, read in the 5′→3′ direction, is the same on both strands of DNA). However, a few of them fall slightly outside this norm. Table 7-2 lists the sequences recognized by a few Type II restriction endonucleases.
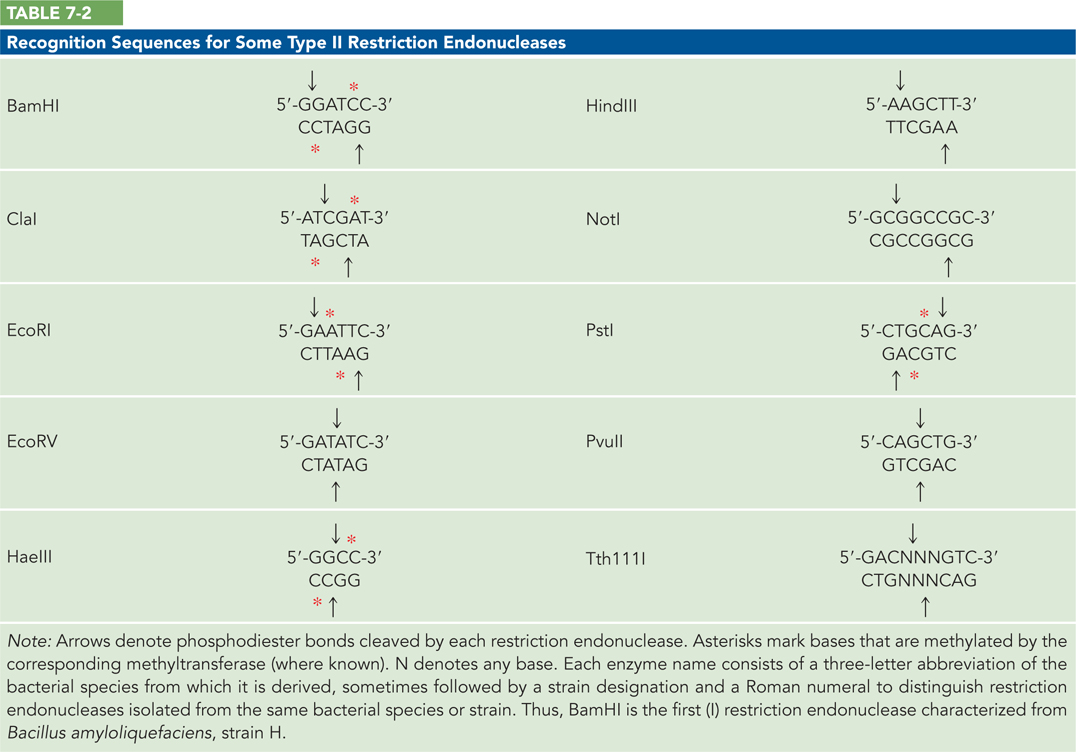
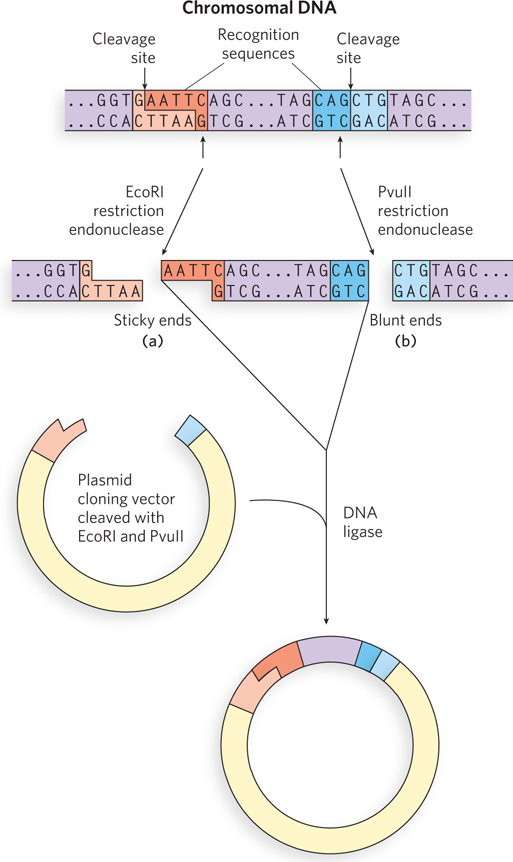
Some restriction endonucleases make staggered cuts across the two DNA strands, leaving 2 to 4 nucleotides of one strand unpaired at each resulting end. Depending on which restriction enzyme is used, cleavage might occur such that the extended strand has either a 5′ or a 3′ end (called a 5′ or 3′ overhang). These unpaired strands are referred to as sticky ends, because they can base-
The average size of the DNA fragments produced by cleaving genomic DNA with a restriction endonuclease depends on the frequency with which a particular recognition sequence occurs in the DNA molecule; this in turn depends largely on the length of the recognition sequence. In a DNA molecule with a random sequence in which all four nucleotides are equally abundant, a 6 bp sequence recognized by a restriction endonuclease would occur, on average, once every 46 (4,096) bp. A 4 bp recognition sequence would occur much more often, about once every 44 (256) bp. In laboratory experiments, the fragment size can be increased by terminating the reaction before completion—
215
Other ways to obtain fragments of DNA for cloning are nonspecific shearing of the DNA, synthesis of the desired fragment, or use of the polymerase chain reaction (PCR). Many protocols are used to shear DNA including sonication, which uses sound energy to bring about hydrodynamic shearing, or simply forcing long DNA strands through a fine-
After a target DNA fragment is obtained, DNA ligase can be used to join it to a cloning vector. The ligation reaction is greatly facilitated if the ends to be joined (ligated) have complementary sticky ends, as was apparent in the earliest recombinant DNA experiments (see the How We Know section at the end of this chapter). This is normally accomplished by cleaving the vector DNA with the same restriction enzyme used to prepare the target DNA fragments. DNA ligase catalyzes the formation of a phosphodiester bond between a 3′ hydroxyl at the end of one DNA strand and a 5′ phosphate at the end of another strand (see Figure 5-12).
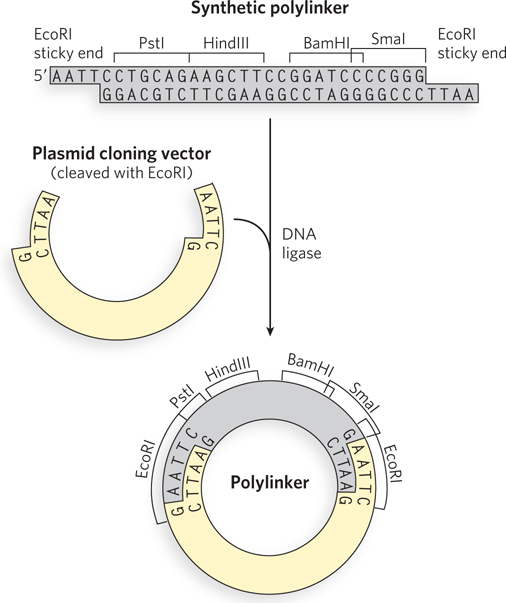
Researchers can create new DNA sequences by inserting synthetic DNA fragments, called linkers, between the ends that are being ligated. Inserted DNA fragments with multiple recognition sequences for restriction endonucleases (often useful later in the experiment as points for inserting additional DNA by cleavage and ligation) are known as polylinkers (Figure 7-3).
Cloning Vectors Allow Amplification of Inserted DNA Segments
Genes or genomic segments are cloned for many different reasons. This is reflected in the use of a large variety of cloning vectors. The principles that govern the delivery of recombinant DNA in clonable form to a host cell, and its subsequent amplification in the host, are well illustrated by considering some popular cloning vectors used in experiments with E. coli and yeast: plasmids, bacterial artificial chromosomes, and yeast artificial chromosomes. Modern cloning vectors provide an array of options, allowing an investigator to tailor the cloning exercise to a particular goal: DNA sequencing, gene expression for protein purification, study of the effects of mutations, or creation of many kinds of gene alterations.
216
Plasmids A plasmid is a circular DNA molecule that replicates separately from the host chromosome. The wide variety of naturally occurring bacterial plasmids range in size from 5,000 to 400,000 bp. Many of the plasmids found in bacterial populations are little more than molecular parasites, similar to viruses but with a more limited capacity to transfer from one cell to another. To survive in the host cell, plasmids contain or incorporate several specialized sequences that enable them to use the cell’s resources for their own replication and gene expression.
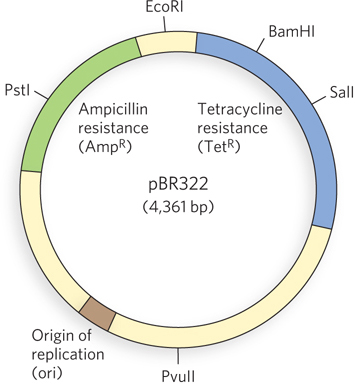
Naturally occurring plasmids usually have a symbiotic role in the cell. They may provide genes that confer resistance to antibiotics or that perform new functions for the cell. For example, the Ti plasmid of Agrobacterium tumefaciens allows the host bacterium to colonize plant cells and make use of the plant’s resources. The same properties that enable plasmids to grow and survive in a bacterial or eukaryotic host are useful to researchers who want to engineer a vector for cloning a specific DNA segment. The classic E. coli plasmid pBR322, constructed in 1977, is a good example of a plasmid with features useful in almost all cloning vectors (Figure 7-4):
The plasmid pBR322 has an origin of replication, or ori: a sequence where replication is initiated by cellular enzymes (see Chapter 11). This sequence is required to propagate the plasmid. An associated regulatory system is present that limits replication to maintain pBR322 at a level of 10 to 20 copies per cell.
217
The plasmid contains genes that confer resistance to the antibiotics tetracycline (TetR) and ampicillin (AmpR), allowing the selection of cells that contain the intact plasmid or a recombinant version of the plasmid (discussed below).
Several unique recognition sequences in pBR322 are targets for restriction endonucleases (PstI, EcoRI, BamHI, SalI, and PvuII), providing sites where the plasmid can be cut to insert foreign DNA.
The small size of the plasmid (4,361 bp) facilitates both its entry into cells and the biochemical manipulation of the DNA. This small size is generated simply by trimming away many DNA segments from a larger, parent plasmid—
sequences that the molecular biologist does not need.
Many variations and enhancements of these basic features of a cloning vector now exist. The replication origins inserted in common plasmid vectors were originally derived from naturally occurring plasmids. Each of these origins is regulated to maintain a particular number of plasmid copies in a cell (the plasmid copy number). Depending on the origin used, the plasmid copy number can vary from one to hundreds or thousands per cell, providing many options for investigators. Two different plasmids cannot function in the same cell if they use the same origin of replication, because the regulation of one will interfere with the replication of the other. Such plasmids are said to be incompatible. When a researcher wants to introduce two or more different plasmids into a bacterial cell, each plasmid must have a different replication origin.
In the laboratory, small plasmids can be introduced into bacterial cells by a process called transformation. The cells (often E. coli, but other bacterial species are also used) and plasmid DNA are incubated together at 0°C in a calcium chloride solution, then subjected to heat shock by rapidly shifting the temperature to between 37°C and 43°C. For reasons not well understood, some of the cells treated in this way take up the plasmid DNA. Some species of bacteria, such as Acinetobacter baylyi, are naturally competent for DNA uptake and do not require the calcium chloride–
Regardless of the approach, relatively few cells take up the plasmid DNA, so a method is needed to identify those that do. The usual strategy is to utilize one of two types of genes in the plasmid, referred to as selectable and screenable markers. Selectable markers either permit the growth of a cell (positive selection) or kill the cell (negative selection) under a defined set of conditions. The plasmid pBR322 provides opportunities for both positive and negative selection (Figure 7-5). A screenable marker is a gene encoding a protein that causes a visible change in cell appearance, such as producing a color or making the cell fluoresce. Cells are not harmed whether the gene is present or not. The cells that carry the recombinant plasmid are easily identified by the colored or fluorescent colonies they produce.
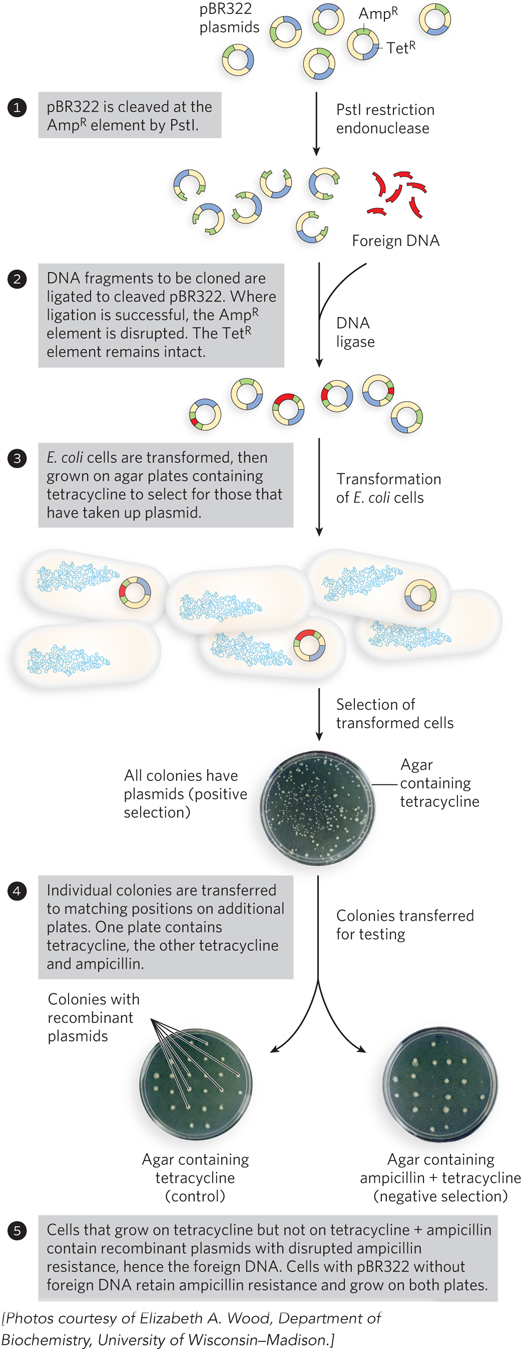
Transformation of typical bacterial cells with purified DNA (never a very efficient process) becomes less successful as plasmid size increases, and it is difficult to clone DNA segments longer than about 15,000 bp when plasmids are used as the vector.
To illustrate the use of a plasmid as a cloning vector, consider a typical bacterial gene that encodes a recombinase called the RecA protein (see Chapter 13). In most bacteria, the gene encoding RecA is one of thousands of other genes on a chromosome millions of base pairs long. The recA gene is just over 1,000 bp long. A plasmid would be a good choice for cloning a gene of this size. As described later, the cloned gene can be altered in a variety of ways, and the gene variants can be expressed at high levels to enable purification of the encoded proteins.
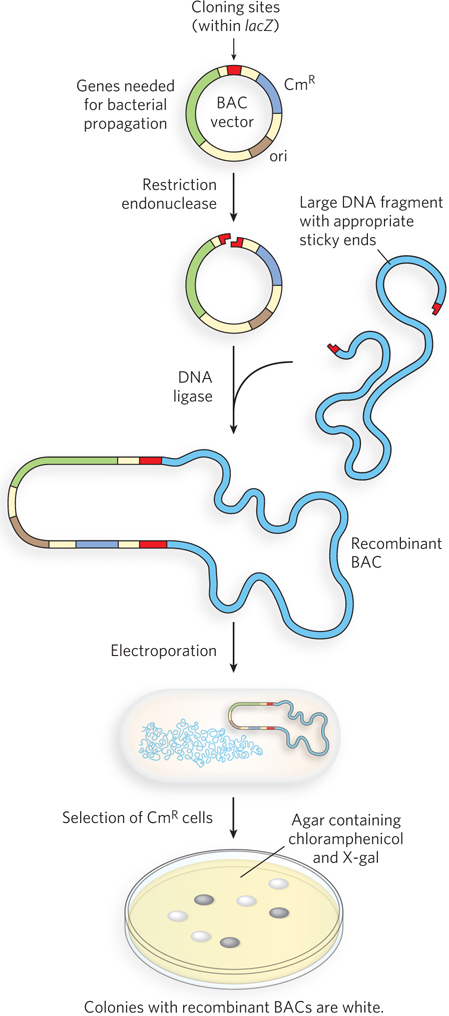
Bacterial Artificial Chromosomes Large genome sequencing projects often require the cloning of much longer DNA segments than can typically be incorporated into standard plasmid cloning vectors such as pBR322. To meet this need, plasmid vectors have been developed with special features that allow the cloning of very long segments (typically 100,000 to 300,000 bp) of DNA. Once such large segments of cloned DNA have been added, these vectors are large enough to be thought of as chromosomes and are known as bacterial artificial chromosomes, or BACs (Figure 7-6).
218
219
A BAC vector is a relatively simple plasmid, generally not much larger than other plasmid vectors. To accommodate very long segments of cloned DNA, BAC vectors have stable origins of replication that maintain the plasmid at one or two copies per cell. The low copy number is useful in cloning large segments of DNA because it limits the opportunities for unwanted recombination reactions that can unpredictably alter large cloned DNAs over time. BACs also include par genes, which encode proteins that direct the reliable distribution of the recombinant chromosomes to daughter cells at cell division, thereby increasing the likelihood of each daughter cell carrying one copy, even when few copies are present. The BAC vector includes both selectable and screenable markers. The BAC vector shown in Figure 7-6 contains a gene for resistance to the antibiotic chloramphenicol (CmR). Positive selection for vector-
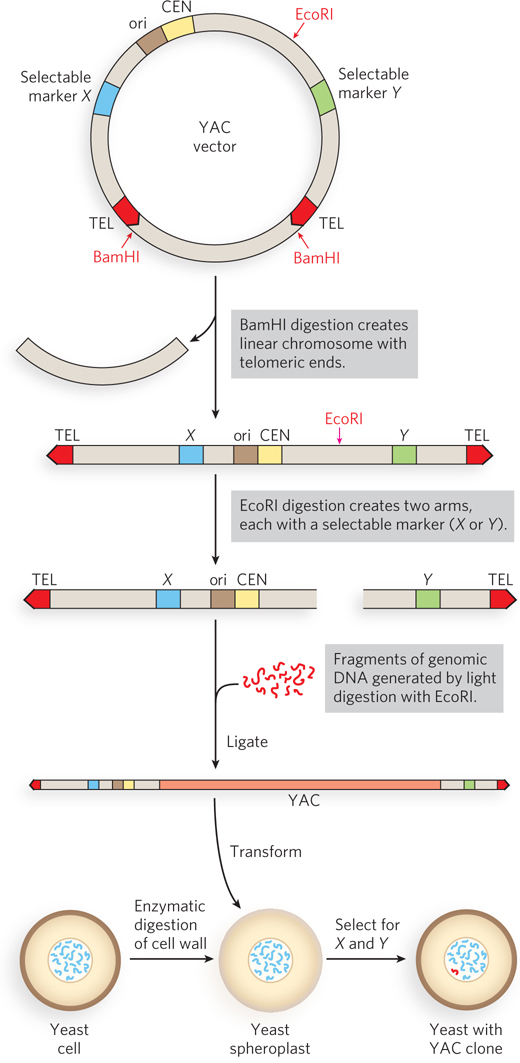
Yeast Artificial Chromosomes As with E. coli, yeast genetics is a well-
Research on large genomes and the associated need for high-
220
The genomic DNA to be cloned is prepared by partial digestion with restriction endonucleases to obtain a suitable fragment size. Genomic fragments are then separated by pulsed field gel electrophoresis, a variation of gel electrophoresis that segregates very large DNA segments. DNA fragments of appropriate size (up to about 2 × 106 bp) are mixed with the prepared vector arms and ligated. The ligation mixture is then used to transform yeast cells (pretreated to partially degrade their cell walls) with these very large DNA molecules—
As with BACs, YAC vectors can be used to clone very long segments of DNA. In addition, the DNA cloned in a YAC can be altered to study the function of specialized sequences in chromosome metabolism, mechanisms of gene regulation and expression, and many other problems in eukaryotic molecular biology.
DNA Libraries Provide Specialized Catalogs of Genetic Information
A DNA library is a collection of DNA clones, gathered together for purposes of genome sequencing, gene discovery, or determination of gene function. The library can take a variety of forms, depending on the source of the DNA and the ultimate purpose of the library.
One of the largest is a genomic library, produced when the complete genome of an organism is cleaved into thousands of fragments and all the fragments are cloned by insertion into a cloning vector. Building such a library has traditionally been a prelude to large sequencing projects. The first step is partial digestion of the DNA by restriction endonucleases, such that any given sequence will appear in fragments of a range of sizes—
With the increasing availability of genome sequences, the utility of genomic libraries is diminishing, and investigators are building more specialized libraries for studying gene function. An example is a library that includes only those sequences of DNA that are expressed—transcribed into RNA—
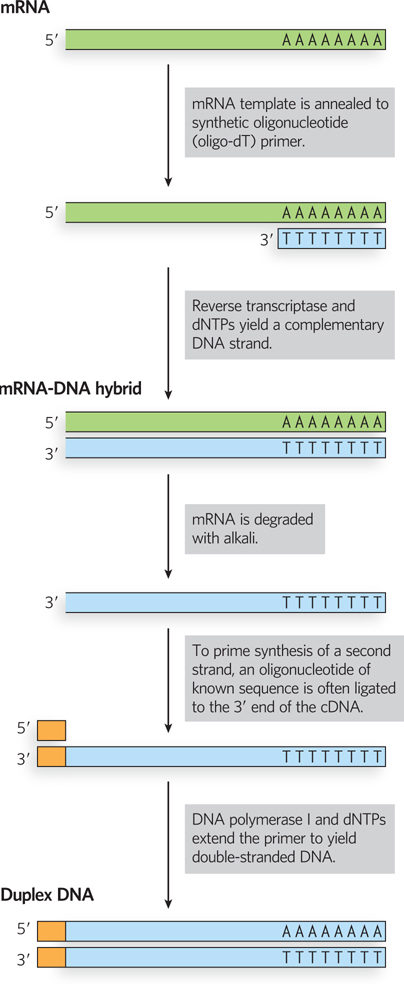
The search for a particular gene is made easier by focusing on a cDNA library generated from the mRNAs of a cell known to express that gene. For example, if we wished to clone globin genes, we could first generate a cDNA library from erythrocyte precursor cells, in which about half the mRNAs code for globins. A particular gene or gene segment in a library can be detected by the hybridization techniques introduced in Chapter 6. If a researcher knows something about the sequence of the DNA being sought, a short nucleic acid complementary to that sequence can be synthesized, labeled, and used to identify cells carrying a recombinant plasmid that incorporates that particular sequence.
221
SECTION 7.1 SUMMARY
Genes are isolated for study by cloning them into vectors that permit their selection and amplification. A gene or genomic segment is cut out of a chromosome with a restriction enzyme and ligated into a vector. The recombinant vector is transferred into a host cell and is amplified in this transformed cell.
Gene cloning relies on an arsenal of enzymes made available by advances in molecular biology, including restriction endonucleases, DNA ligase, DNA polymerase, and reverse transcriptase.
Important cloning vectors include plasmids, bacterial artificial chromosomes, and yeast artificial chromosomes. BACs and YACs allow the cloning of very long DNA segments.
DNA libraries are specialized archives used in gene sequencing, gene discovery, or the functional characterization of proteins.