7.3 UNDERSTANDING THE FUNCTIONS OF GENES AND THEIR PRODUCTS
One of the challenges in molecular biology is to identify the functions of the myriad genes being discovered in large genome sequencing projects, which we survey in Chapter 8. When the complete sequence of an organism’s genome becomes available, we often lack functional information for half or more of the defined genes. Biotechnology provides some shortcuts to understanding the functions of gene products, and we describe some of the key technologies here (and expand on their application in Chapter 8). To begin to define the function of a new, previously unstudied protein, a molecular biologist gathers clues—defining the protein’s location in cells, what it interacts with, when it is expressed, and what happens to the organism when it is absent. These clues, together with information about the activity of the purified protein in vitro, eventually paint a consistent functional picture.
Protein Fusions and Immunofluorescence Can Localize Proteins in Cells
Often, an important clue to a gene product’s function comes from determining its location within the cell. For example, a protein found exclusively in the nucleus could be involved in processes that are unique to that organelle, such as transcription, replication, or chromatin condensation. Researchers often engineer fusion proteins for the purpose of locating a protein in the cell or organism. Some of the most useful fusions involve the addition of marker proteins that allow the investigator to determine the location by direct visualization or by immunofluorescence.
A particularly useful marker is the gene for green fluorescent protein (GFP). A target gene (coding the protein of interest) fused to the GFP gene generates a fusion protein that is highly fluorescent—it literally lights up when exposed to blue light—and can be visualized directly in a living cell. GFP is a protein derived from the jellyfish Aequorea victoria (see the How We Know section at the end of this chapter). It has a β-barrel structure (Figure 7-22a), and the fluorophore (the fluorescent component of the protein) is in the center of the barrel. The fluorophore is derived from a rearrangement and oxidation of several amino acid residues. Because this reaction is autocatalytic and requires no other proteins or cofactors (other than molecular oxygen), GFP is readily cloned in an active form in almost any cell. Just a few molecules of this protein can be observed microscopically, allowing the study of its location and movements in a cell. Careful protein engineering, coupled with the isolation of related fluorescent proteins from other marine coelenterates, has made a wide range of these proteins available, with an array of colors (Figure 7-22b) and other characteristics (brightness, stability). With this technology, for example, the protein GLR1 (a glutamate receptor of nervous tissue) has been visualized as a GLR1-GFP fusion protein in the nematode Caenorhabditis elegans (Figure 7-22c).
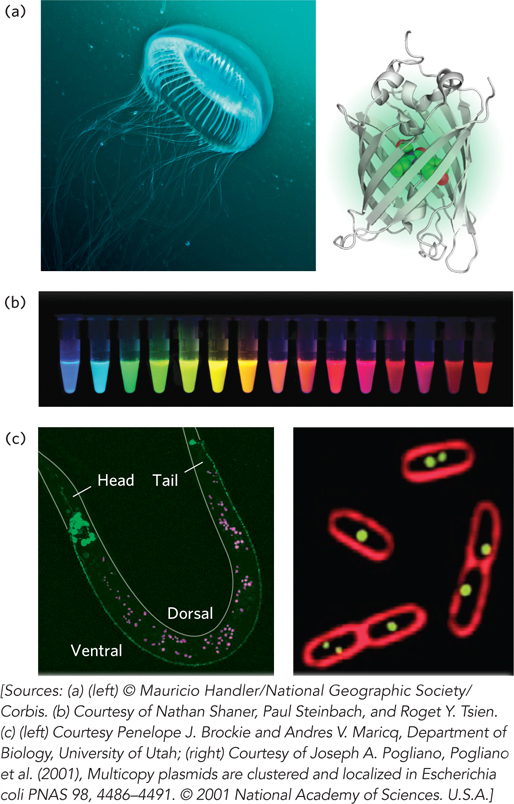
Figure 7-22: Green fluorescent protein (GFP). (a) The source of GFP is the jellyfish Aequorea victoria (left); the bioluminescent photo-organs are visible. The GFP protein has a β-barrel structure (right); the fluorophore (red) is in the center of the barrel. (b) Variants of GFP are now available in almost any color of the visible spectrum. (c) Caenorhabditis elegans, a nematode worm (left), containing a GLR1-GFP fusion protein. GLR1 is a glutamate receptor of nervous tissue. In the E. coli cells (right), the membranes are stained with a red fluorescent dye. The cells are expressing a protein that binds to a resident plasmid, fused to GFP. The green spots indicate the locations of plasmids.
In many cases, visualization of a GFP fusion protein in a live cell is not possible, or is not practical or desirable. The GFP fusion protein may be inactive or may not be expressed at sufficient levels to allow visualization. In this case, immunofluorescence is an alternative approach for visualizing the endogenous (unaltered) protein, although fusion proteins are sometimes used here, too. This approach requires the fixation (and thus death) of the cell. The protein of interest is expressed either unaltered or as a fusion protein with an epitope tag, a short protein sequence that is bound tightly by a well-characterized, commercially available antibody. Fluorescent molecules (fluorochromes) are attached to this antibody.
More commonly, a second antibody is added that binds specifically to the first (primary) one, and it is the secondary antibody that has the attached fluorochrome(s) (Figure 7-23). A variation of this indirect visualization approach is to attach biotin molecules to the primary antibody, then add streptavidin (a bacterial protein closely related to avidin, biotin’s natural ligand) that is complexed with fluorochromes. The interaction between biotin and streptavidin is one of the strongest and most specific known, and the potential to add multiple fluorochromes to each target protein gives this method great sensitivity.
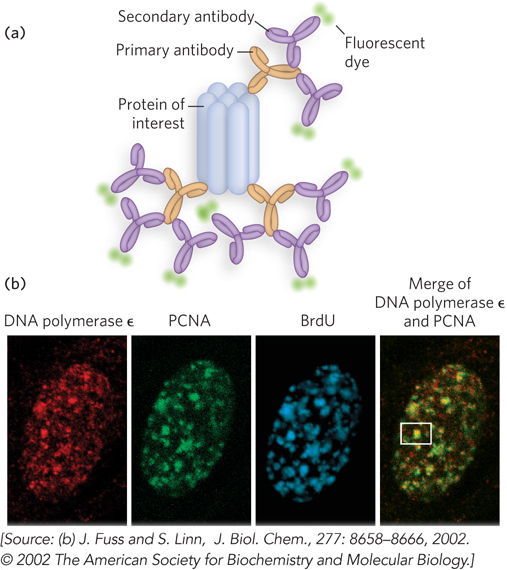
Figure 7-23: Indirect immunofluorescence. (a) The protein of interest is bound to a primary antibody, and a secondary antibody is added; this secondary antibody, with one or more attached fluorescent groups, binds to the primary antibody. Multiple secondary antibodies can bind the primary antibody, amplifying the signal. If the protein of interest is in the interior of the cell, the cell is fixed and permeabilized, and the two antibodies are added in succession. (b) The end result is an image in which bright spots indicate the location of the protein of interest in the cell. The images here show a nucleus from a human fibroblast, stained with antibodies and fluorescent labels for DNA polymerase ε, for PCNA, an important polymerase accessory protein, and for bromo-deoxyuridine (BrdU), a nucleotide analog. The BrdU identifies regions undergoing active DNA replication. The patterns of staining show that DNA polymerase ε and PCNA colocalize to regions of active DNA synthesis, with one example marked by a white box.
Highly specialized cDNA libraries (see Figure 7-8) can be made by cloning cDNAs or cDNA fragments into a vector that fuses each cDNA sequence with the sequence for a marker, also called a reporter gene. For example, libraries have been developed in which all the genes in the library are fused to the GFP gene (Figure 7-24). Each cell in the library expresses one of these fused genes. The cellular location of the product of any gene represented in the library can be studied—assuming that the particular fusion protein is expressed at sufficient levels and retains its normal function and location—by examining cells that express the appropriate fused gene, to detect light foci that reveal the protein’s presence.
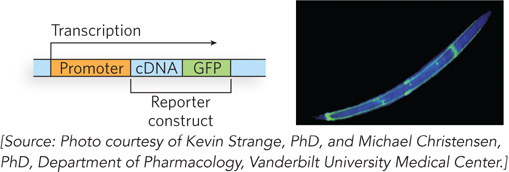
Figure 7-24: Specialized DNA libraries. Cloning of a cDNA next to the GFP gene creates a reporter construct. Transcription proceeds through the gene of interest (the inserted cDNA) and the reporter gene (here, GFP), and the mRNA transcript is expressed as a fusion protein. The GFP part of the protein is visible with the fluorescence microscope. Although only one example is shown, thousands of genes can be fused to GFP in similar constructs and stored in libraries in which each cell or organism in the library expresses a different protein fused to GFP. If the fusion protein is properly expressed, its location in the cell or organism can be assessed. The photograph shows a nematode worm (C. elegans) containing a GFP fusion protein expressed only in the four “touch” neurons that run the length of its body.
Proteins Can Be Detected in Cellular Extracts with the Aid of Western Blots
Western blots, also known as immunoblots, make use of antibodies to detect the presence of specific proteins in a biological sample, such as in a certain tissue or at a given time in an organism’s development (Figure 7-25). The antibodies are obtained by purifying the protein of interest, inoculating a chicken or rabbit with the protein, and isolating the resulting antibodies from the serum. The first few steps are similar to those described for Southern blots (for nucleic acids) in Chapter 6: a protein sample is subjected to electrophoresis in a polyacrylamide gel, and the gel is then blotted on a membrane to which proteins in the gel adhere. The remaining steps of Western blotting are specific for the detection of proteins rather than nucleic acids. The membrane is first washed with a protein solution to coat the membrane, to prevent nonspecific adherence of the antibodies. Then it is washed with a solution containing the protein-specific rabbit or chicken antibodies, which bind to the immobilized target protein. A second solution is then added, containing a second antibody that specifically binds to the first (e.g., an antibody derived from goats that binds all rabbit-derived antibodies of the abundant IgG class of circulating antibodies). The second antibody has an attached radioactive or fluorescent label to allow visualization of protein-antibody complexes. In some cases, a single labeled antibody is used. The procedure is sensitive to changes in the amount of target protein present, so increases or decreases in cellular protein levels are readily monitored.
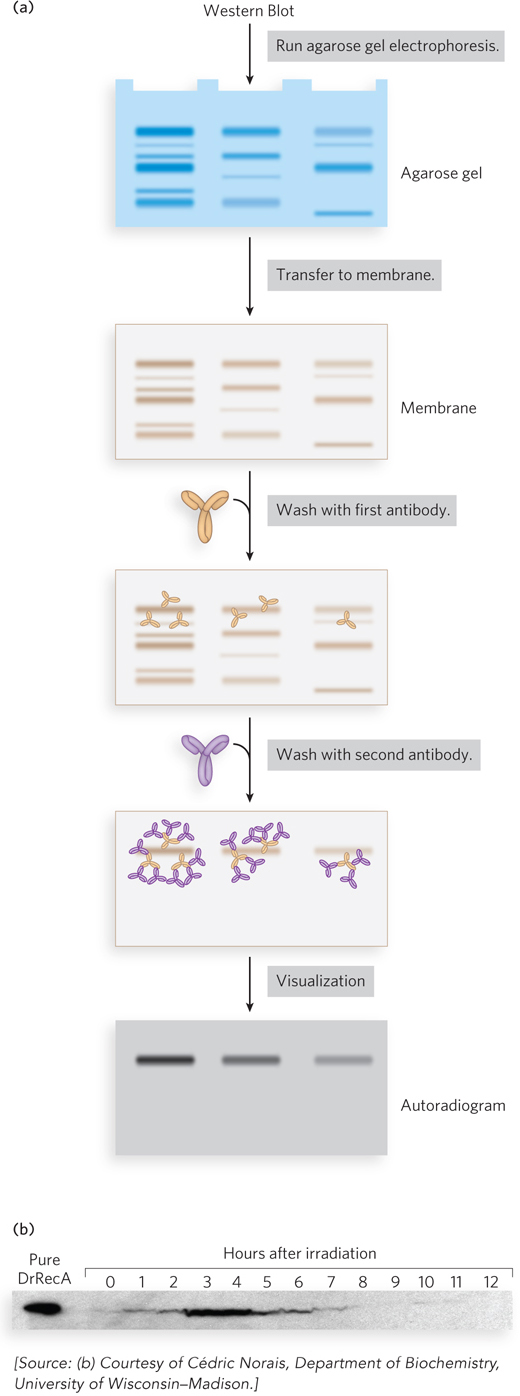
Figure 7-25: Western blots. (a) Proteins are subjected to electrophoresis, then transferred from the gel to a membrane. The membrane is washed successively with the first antibody and the second (labeled) antibody, allowing visualization of the protein of interest. (b) A Western blot shows levels of RecA protein in cells of the highly radiation-resistant bacterium Deinococcus radiodurans. The D. radiodurans protein DrRecA is first induced and then repressed, according to the need for DNA repair, in the hours following high-level irradiation.
Protein-Protein Interactions Can Help Elucidate Protein Function
Another key to defining the function of a particular protein is to determine what it binds to. In the case of protein-protein interactions, the association of a protein of unknown function with a protein having a known function can provide useful and compelling “guilt by association.” The techniques used in this effort are quite varied.
Purification of Protein Complexes With the construction of cDNA libraries in which each gene is fused to an epitope tag, investigators can precipitate the protein product of a gene by complexing it with the antibody that binds the epitope, a process called immunoprecipitation (Figure 7-26). If the tagged protein is expressed in cells, other proteins that bind to it precipitate with it. Identifying the associated proteins reveals some of the intracellular protein-protein interactions of the tagged protein. There are many variations of this process. For example, a crude extract of cells that express a tagged protein is added to a column containing immobilized antibody. The tagged protein binds to the antibody, and proteins that interact with the tagged protein are sometimes also retained on the column. The connection between the protein and the tag is cleaved with a specific protease, and the protein complexes are eluted from the column and analyzed. Researchers can use these methods to define complex networks of interactions within a cell. In principle, the chromatographic approach to analyzing protein-protein interactions can be used with any type of protein tag (His tag, GST, etc.) that can be immobilized on a suitable chromatographic medium.
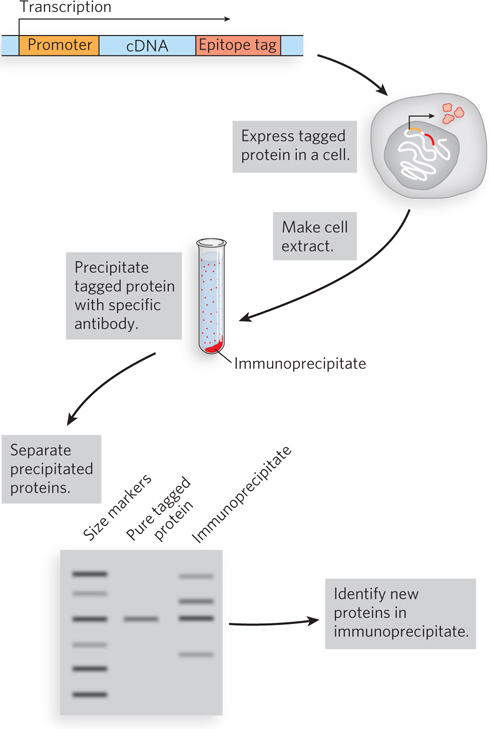
Figure 7-26: The use of epitope tags to study protein-protein interactions. The gene of interest is cloned next to a gene for an epitope tag, and the resulting fusion protein is precipitated by antibodies to the epitope (immunoprecipitation). Any other proteins that interact with the tagged protein also precipitate, thereby helping to elucidate protein-protein interactions.
The selectivity of this approach has been enhanced with the use of tandem affinity purification (TAP) tags. Two consecutive tags, polypeptides known as Protein A and calmodulin-binding peptide, are fused to a target protein, and the fusion protein is expressed in a cell (Figure 7-27). A crude extract containing the TAP-tagged fusion protein is passed through a column matrix that has attached IgG antibodies that bind Protein A. Most of the unbound proteins are washed through the column, but proteins associated with the target protein are retained. Protein A is then cleaved from the fusion protein with the enzyme TEV protease, and the shortened target protein and associated proteins are eluted from the column. The eluent is then passed through a second column containing a matrix of calmodulin beads. Loosely bound proteins are again washed from the column, and the target protein is eluted from the column with its associated proteins. The two consecutive purification steps eliminate any weakly bound contaminating proteins. False positives are minimized, and protein interactions that persist through both steps are likely to be functionally significant.
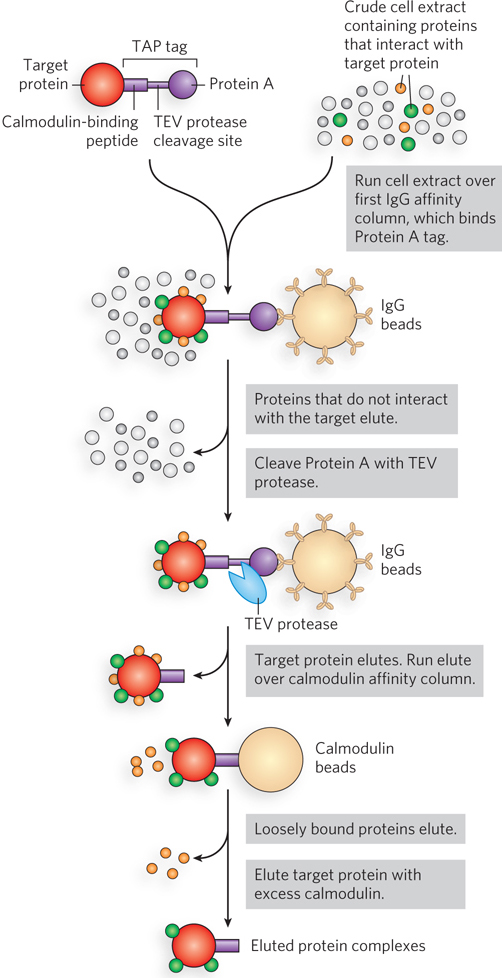
Figure 7-27: Tandem affinity purification (TAP) tags. A TAP-tagged protein and associated proteins are isolated by two consecutive affinity purifications.
Yeast Two-Hybrid and Three-Hybrid Analysis A sophisticated genetic approach to defining protein-protein interactions is based on the properties of the Gal4 protein (Gal4p), which activates the transcription of GAL genes in yeast (genes encoding the enzymes of galactose metabolism; see Chapter 21). Gal4p has two domains: one that binds a specific DNA sequence and another that activates RNA polymerase to synthesize mRNA from an adjacent gene. The two domains of Gal4p are stable when separated, but activation of RNA polymerase requires interaction with the activation domain, which in turn requires positioning by the DNA-binding domain. Hence, the domains must be brought together to function correctly.
In yeast two-hybrid analysis, the protein-coding regions of the genes to be analyzed are fused to the yeast gene for either the DNA-binding domain or the activation domain of Gal4p, and the resulting genes express a series of fusion proteins (Figure 7-28). If a protein fused to the DNA-binding domain interacts with a protein fused to the activation domain, transcription is activated. The reporter gene transcribed by this activation is generally one that yields a protein required for growth or an enzyme that catalyzes a reaction with a colored product. Thus, when grown on the proper medium, cells that contain a pair of interacting proteins are easily distinguished from those that do not. A library can be set up with a particular yeast strain in which each cell in the library has a gene fused to the Gal4p DNA-binding domain gene, and many such genes are represented in the library. In a second yeast strain, a gene of interest is fused to the gene for the Gal4p activation domain. The yeast strains are mated, and individual diploid cells are grown into colonies. This allows large-scale screening for cellular proteins that interact with the target protein.
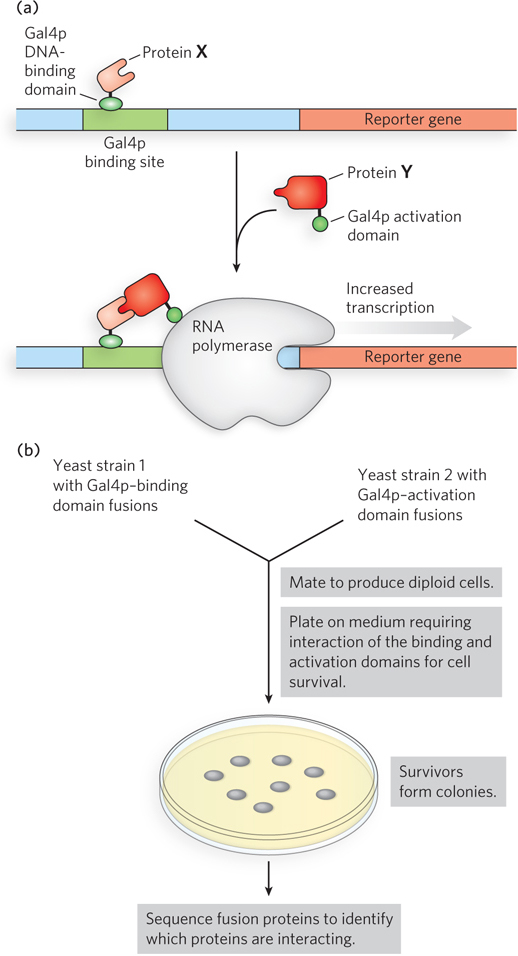
Figure 7-28: Yeast two-hybrid analysis. (a) The goal is to bring together the DNA-binding domain and the activation domain of the yeast Gal4 protein (Gal4p) through the interaction of two proteins, X and Y, to which one or other of the domains is fused. This interaction is accompanied by the expression of a reporter gene. (b) The two gene fusions are created in separate yeast strains, which are then mated. The mated mixture is plated on a medium on which the yeast cannot survive unless the reporter gene is expressed. Thus, all surviving colonies have interacting fusion proteins. Sequencing of the fusion proteins in the survivors reveals which proteins are interacting.
The function of many key regulatory proteins, especially in eukaryotic cells, involves specific interactions between the proteins and RNA molecules. A strategy called yeast three-hybrid analysis has been developed to screen for protein-RNA interactions (Figure 7-29). For a known RNA-binding protein, this method yields a rapid identification of all or most of the RNAs that the protein binds. The method uses three engineered elements: two fusion proteins and a plasmid library. The fusion proteins are (1) the protein of interest fused to the Gal4p transcription-activation domain, and (2) the DNA-binding domain of a protein known as LexA fused to an RNA-binding protein called MS2. The LexA portion binds to a specific DNA sequence, and MS2 binds tightly to an RNA hairpin with a defined sequence. The DNA-binding site for the LexA protein is placed upstream of a reporter gene. The third element, the plasmid library, consists of the gene encoding the RNA hairpin recognized by MS2, fused to random sequences. When transcribed, each MS2 RNA is fused to another RNA segment. If a particular expressed RNA is bound by the protein of interest, the RNA serves as a tether, linking the first fusion protein with the second and activating transcription of the reporter gene. With this method, a few dozen RNA molecules that specifically bind the target protein can be isolated from a library containing millions of cloned RNAs.
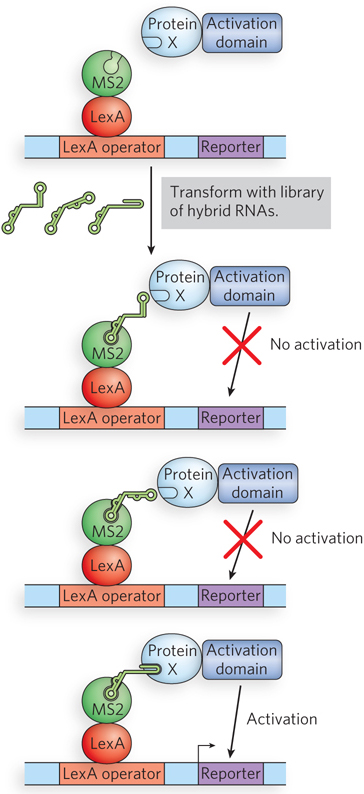
Figure 7-29: Yeast three-hybrid analysis. Two fusion proteins bind simultaneously to a hybrid RNA molecule to permit expression of a reporter gene. An RNA library consisting of random-sequence RNA segments fused to a hairpin recognized by MS2 is screened. If the protein of interest (X) binds the random RNA sequence expressed in a given cell, that cell will survive and produce a colony.
These techniques for determining cellular localization and molecular interactions provide important clues to protein function. However, they do not replace classical biochemistry and molecular biology. They simply give researchers an expedited entrée into important new biological problems. When paired with the simultaneously evolving tools of biochemistry and molecular biology, the techniques described here are speeding the discovery not only of new proteins, but of new biological processes and mechanisms.
DNA Microarrays Reveal Cellular Protein Expression Patterns and Other Information
Major refinements of the technology underlying DNA libraries, PCR, and hybridization have come together in the development of DNA microarrays, which allow the rapid and simultaneous screening of many thousands of genes. In the most commonly used technique, DNA segments from genes of known sequence, a few dozen to hundreds of base pairs long, are synthesized directly on a solid surface by a process called photolithography (Figure 7-30). Thousands of independent sequences are generated, each occupying a tiny part, or spot, of a surface measuring just a few square centimeters. The pattern of sequences is predesigned, with each of many thousands of spots containing sequences derived from a particular gene. The resulting array, or chip, may include sequences derived from every gene of a bacterial or yeast genome, or selected families of genes from a larger genome. Once constructed, the microarray can be probed with mRNAs or cDNAs from a particular cell type or cell culture to identify the genes being expressed in those cells.
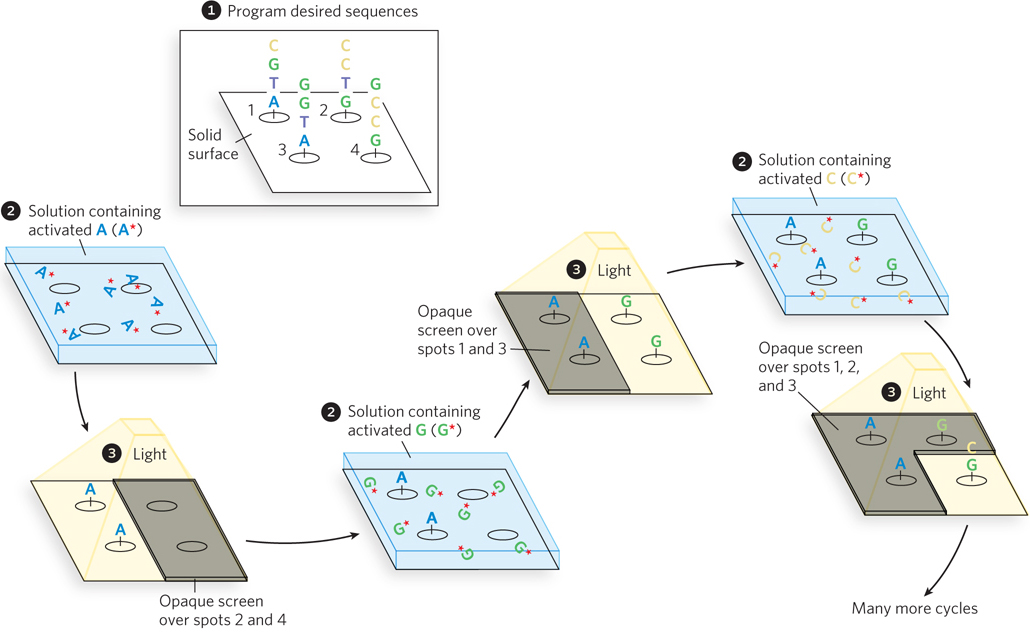
Figure 7-30: Photolithography to create a DNA microarray.  A computer is programmed with the desired oligonucleotide sequences.
A computer is programmed with the desired oligonucleotide sequences.  The reactive groups, attached to a solid surface, are initially rendered inactive by photoactive blocking groups, which can be removed by a flash of light. An opaque screen blocks the light from certain groups, preventing their activation. Other areas or “spots” are exposed.
The reactive groups, attached to a solid surface, are initially rendered inactive by photoactive blocking groups, which can be removed by a flash of light. An opaque screen blocks the light from certain groups, preventing their activation. Other areas or “spots” are exposed.  A solution containing one activated nucleotide (e.g., A*) is washed over the spots. The 5′ hydroxyl of the nucleotide is blocked to prevent unwanted reactions, and the nucleotide links to the surface groups at the appropriate spots through its 3′ hydroxyl. The surface is washed successively with solutions containing each remaining activated nucleotide (G*, C*, T*). The 5′-blocking groups on each nucleotide limit the reactions to addition of one nucleotide at a time, and these groups can also be removed by light. Once each spot has one nucleotide, a second nucleotide can be added to extend the nascent oligonucleotide at each spot, using screens and light to ensure that the correct nucleotides are added at each spot in the correct sequence. This continues until the required sequences are built up on each spot on the surface.
A solution containing one activated nucleotide (e.g., A*) is washed over the spots. The 5′ hydroxyl of the nucleotide is blocked to prevent unwanted reactions, and the nucleotide links to the surface groups at the appropriate spots through its 3′ hydroxyl. The surface is washed successively with solutions containing each remaining activated nucleotide (G*, C*, T*). The 5′-blocking groups on each nucleotide limit the reactions to addition of one nucleotide at a time, and these groups can also be removed by light. Once each spot has one nucleotide, a second nucleotide can be added to extend the nascent oligonucleotide at each spot, using screens and light to ensure that the correct nucleotides are added at each spot in the correct sequence. This continues until the required sequences are built up on each spot on the surface.
A microarray can provide a snapshot of all the genes in an organism, informing the researcher about the genes that are expressed at a given stage in the organism’s development or under a particular set of environmental conditions. For example, the total complement of mRNA can be isolated from cells at two different stages of development and converted to cDNA with reverse transcriptase. With the use of fluorescently labeled deoxyribonucleotides, the two cDNA samples can be made so that one fluoresces red, the other green (Figure 7-31). The cDNA from the two samples is mixed and used to probe the microarray. Each cDNA anneals to only one spot, corresponding to the gene encoding the mRNA that gave rise to that cDNA. Spots that fluoresce green represent genes that produce mRNAs at higher levels at one developmental stage; those that fluoresce red represent genes expressed at higher levels at another stage. If a gene produces mRNAs that are equally abundant at both stages of development, the corresponding spot fluoresces yellow. By using a mixture of two samples to measure relative rather than absolute sequence abundance, the method corrects for inconsistencies among spots in the microarray. The spots that fluoresce provide a snapshot of all the genes being expressed in the cells at the moment they were harvested—gene expression examined on a genome-wide scale. For a gene of unknown function, the time and circumstances of its expression can provide important clues about its role in the cell.
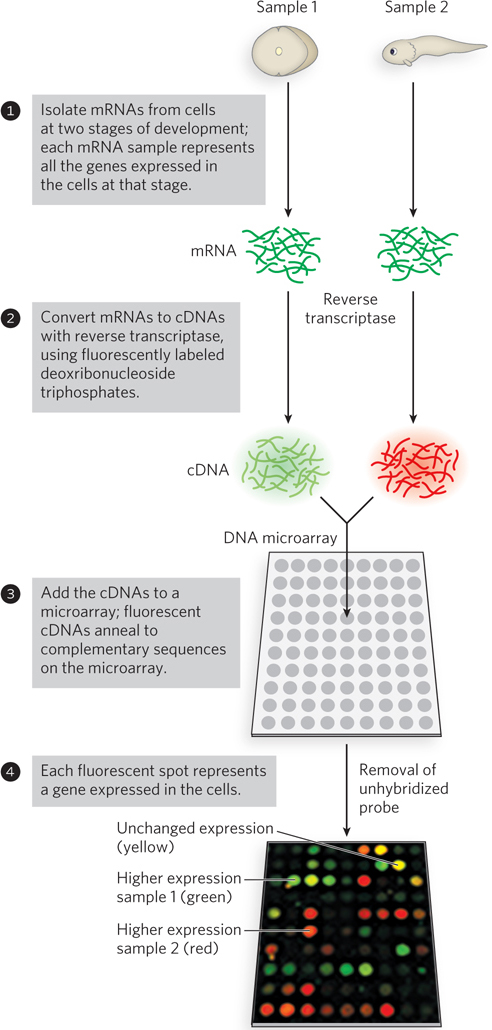
Figure 7-31: A DNA microarray experiment. A microarray can be prepared from any known DNA sequence, from any source. Once the DNA is attached to a solid support, the microarray can be probed with other, fluorescently labeled nucleic acids. Here, mRNA samples are collected from frog cells at two different stages of development: single-cell stage (sample 1) and a later stage (sample 2). The cDNA probes are synthesized with nucleotides that fluoresce in different colors for each sample; a mixture of the cDNAs is used to probe the microarray. The probes anneal to spots containing complementary DNA; if the spot lights up, the corresponding gene is represented in the pool of mRNA used to produce the probes. Green spots represent mRNAs more abundant at the single-cell stage; red spots, sequences more abundant later in development. The yellow spots indicate approximately equal abundance at both stages.
A Gene’s Function Can Be Elucidated by Examining the Effects of Its Absence
One of the most informative paths to understanding the function of a gene is to change (mutate) it or delete it. The investigator can then examine the effects of the genomic alteration on cell growth or function. The methods available to modify genomes grow more sophisticated every year. One increasingly common strategy is to cut the gene at a site that is functionally critical, generating a double-strand break. In eukaryotes, such breaks are most commonly repaired by cellular systems that promote nonhomologous end joining (NHEJ), a process described in Chapter 13. NHEJ seals the double-strand break, but the process is imprecise. Nucleotides are often deleted or added during repair, inactivating the gene. In bacteria, introduced double-strand breaks are usually repaired more accurately by homologous recombination systems (see Chapter 13), but inactivating mutations can appear. Three significant technologies have been developed to cut a gene at a particular site in vivo: zinc finger nucleases, TALENs, and CRISPR/Cas systems.
Zinc finger nucleases (ZFNs) represent a kind of designer DNA cleavage system. A zinc finger (which we encountered in Chapter 4 and is described in detail in Chapter 19) is a protein domain consisting of about 30 amino acid residues. It folds into a characteristic β-α-β structure, aided by a bound Zn2+ ion (Figure 7-32a). Particular amino acid residues in the α helix contact about three consecutive nucleotides in the major groove of a DNA molecule, leading to binding with a substantial degree of selectivity. Researchers can stitch together several zinc fingers in tandem, creating structures that allow the specific recognition of almost any DNA sequence 9 to 18 bp long. When the zinc fingers are fused to a nonspecific nuclease domain (often derived from an enzyme called FokI) to create a ZFN, the DNA sequence to which the ZFN binds is cleaved at a site adjacent to the recognition sequence of the associated zinc fingers. TALENs are similar, except that the DNA binding is directed by a series of TALE (transcription activator–like effector) domains (Figure 7-32b). These are similar in size to zinc fingers but recognize single base pairs; like zinc fingers, they can be linked together and fused to a nonspecific nuclease domain to yield a TALE nuclease, or TALEN. These enzymes can be expressed in a cell, and the resulting enzyme cleaves the target site in the genome to generate a double-strand break. TALENs can be designed to inactivate genes and even to inactivate viral DNA that is integrated into a genome. One drawback is cost: targeting a new DNA sequence necessitates the design, construction, and testing of a new TALEN enzyme, an expensive process that can take weeks or months.
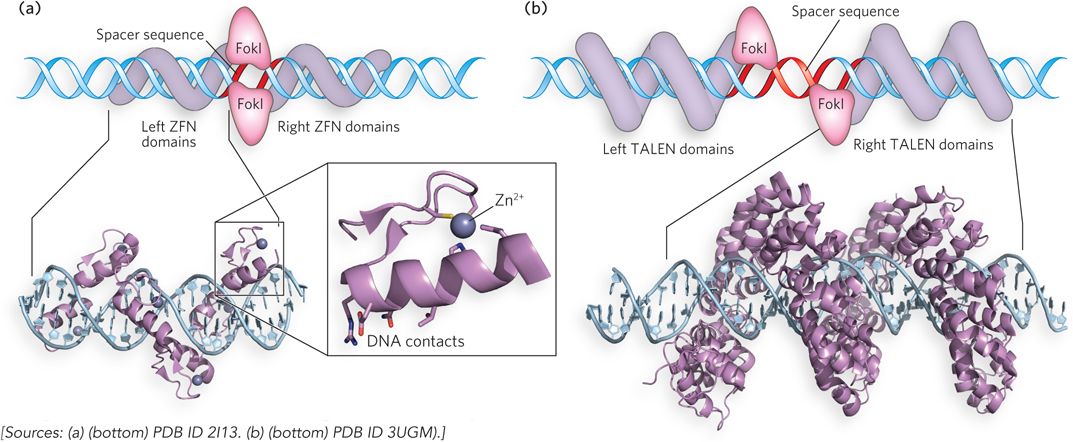
Figure 7-32: Designer nucleases. (a) An engineered zinc finger nuclease (ZFN) in complex with its target DNA (bottom). Surface residues that contact DNA are shown as sticks. Each zinc finger domain contacts 3 or 4 bp in the major groove of the DNA. The complexed Zn2+ ion is shown as a purple ball. The cartoon (top) shows a ZFN dimer bound to DNA. ZFN target sites consist of two zinc finger–binding sites separated by a 5 to 7 bp spacer sequence that is cleaved by the FokI nuclease domain. The half sites recognized by a ZFN need not be identical. (b) A TALE protein bound to its target DNA (bottom). TALEN target sites consist of two TALE-binding sites separated by a spacer sequence of 12 to 20 bp (top).
A more robust and convenient approach to cutting genes in vivo is derived from a kind of bacterial immune system called CRISPR/Cas (clustered, regularly interspaced short palindromic repeats–CRISPR-associated proteins). Spacer sequences inserted between CRISPR repeats, embedded in a bacterial genome, are derived from bacteriophage pathogens. When that bacteriophage attacks a bacterium with the corresponding CRISPR/Cas system, the CRISPR sequences are transcribed to RNA, and individual spacers (plus some adjacent repeat RNA) are cleaved to form products called guide RNAs (gRNAs). A gRNA forms a complex with one or more Cas proteins and, in some cases, with another RNA called a trans-activating CRISPR RNA, or tracrRNA. The resulting complex binds specifically to the invading bacteriophage DNA, cleaving and destroying it with nuclease activities associated with the Cas proteins. A relatively simple example of CRISPR/Cas, found in the bacterium Streptococcus pyogenes, requires only a single Cas protein called Cas9 to recognize and cleave DNA. Work in many laboratories, particularly those of Jennifer Doudna and Emmanuelle Charpentier, has produced a streamlined CRISPR/Cas9 system that is increasingly robust. The gRNA and tracrRNAs are typically fused into a what is called a single guide RNA (sgRNA). The guide sequence can be programmed (by altering its sequence) to target almost any specific genomic site (Figure 7-33). Cas9 has two separate nuclease domains: one domain cleaves the DNA strand paired with the sgRNA, and the other cleaves the opposite DNA strand. Inactivating one domain creates an enzyme that cleaves just one strand, creating a single-strand break, called a nick. The sgRNA is needed both to pair with the target sequence in the DNA and to activate the nuclease domains for cleavage.
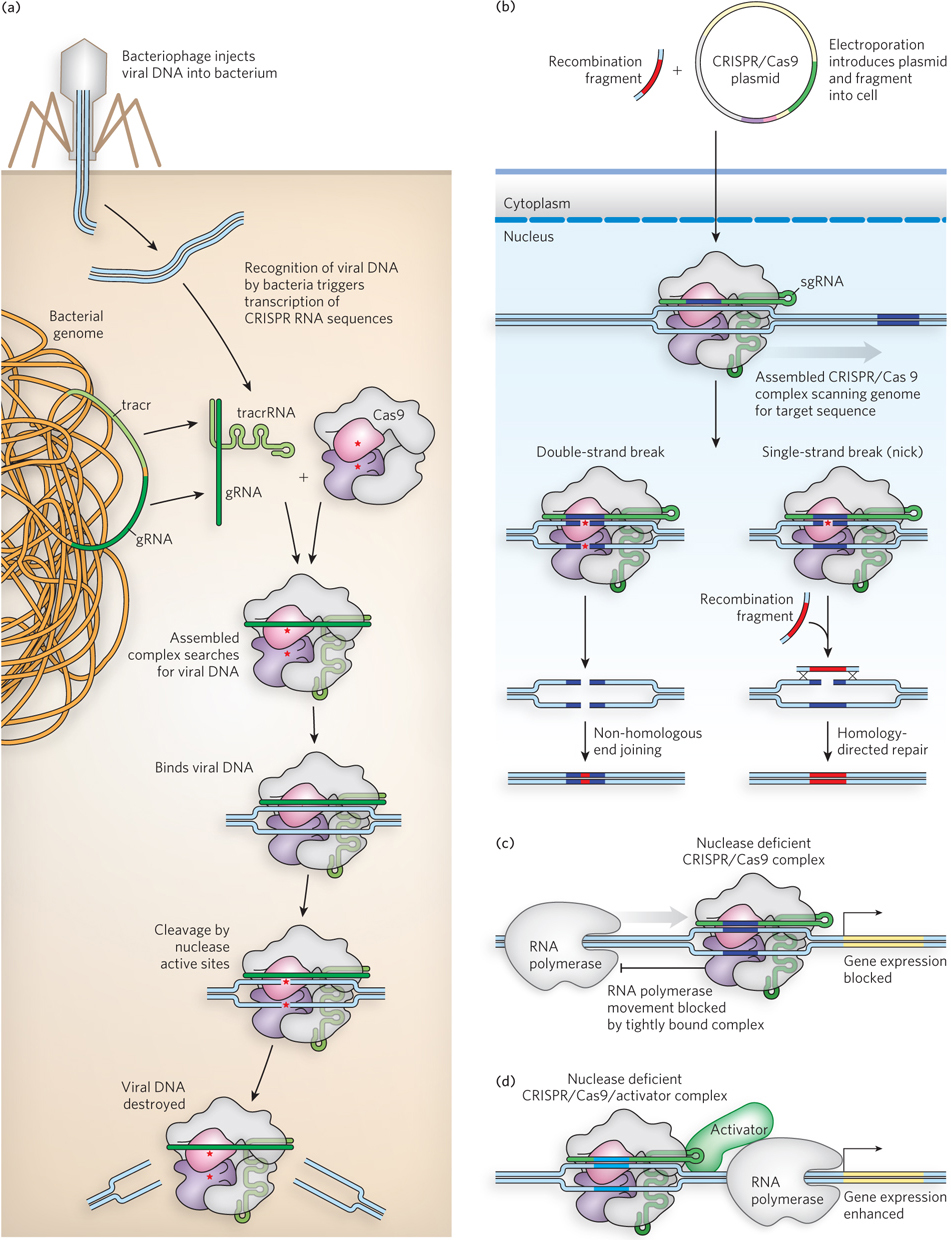
Figure 7-33: The CRISPR/Cas9 system for genomic engineering. (a) A complex consisting of the CRISPR sgRNA and the Cas9 protein binds to a target site in the DNA. Two nuclease active sites in the Cas9 protein are indicated by red asterisks. The cleavage produces a double-strand break or, if one active site is inactivated, a single-strand break (nick). (b) If both nuclease active sites in Cas9 are inactivated, a complex is produced that simply binds tightly to its target sequence. This bound complex can be situated so as to block transcription of a particular gene by RNA polymerase. (c) If both nuclease active sites are inactivated, the Cas9 protein can be fused to a gene activator protein (see Chapter 19). If targeted to a site near an appropriate gene, the complex can be used to trigger enhanced transcription.
Plasmids expressing the required protein and RNA components of CRISPR/Cas9 can be introduced into cells by electroporation (described earlier in this chapter). In cells from many organisms, targeted gene inactivation occurs in 10% to 50% of the treated cells. If a genomic change (mutation) rather than a simple gene inactivation is desired, it can be introduced by recombination when a DNA fragment encompassing the cleavage site and including the planned change enters the cell with the CRISPR/Cas9 plasmids. This recombination is generally more efficient if a nick rather than a double-strand break is introduced at the target site (Figure 7-33a)
Other uses for CRISPR/Cas9 are being developed. If the nuclease active sites of Cas9 are mutationally inactivated, the nuclease-deficient complex still binds tightly to its target. Such a complex can be used to block the movement of RNA polymerase, effectively silencing a gene (Figure 7-33b). In a variation of this strategy, a gene activator (see Chapter 19) can be fused to the nuclease-deficient CRISPR/Cas9. When the resulting complex is targeted to an appropriate genomic site, it can be used to activate transcription of a specific gene of interest (Figure 7-33c). Therapeutic uses of CRISPR/Cas9 are still far in the future, but the potential for alleviating the effects of genetic diseases, HIV, and many other human ailments is enormous.
SECTION 7.3 SUMMARY
The role of a gene of unknown function can be explored by techniques that assess the location of the gene product in the cell, the interactions of the gene product with proteins or RNA, and the expression of the gene under different cellular circumstances.
By fusing a gene of interest with the genes that encode green fluorescent protein or epitope tags, researchers can visualize the cellular location of the gene product, either directly or by immunofluorescence.
The presence of particular proteins in biological samples can be detected with the aid of Western blots.
The interactions of a protein with other proteins or RNA can be investigated with epitope tags and immunoprecipitation or affinity chromatography. Alternatively, interactions in the cell can be probed in yeast two-hybrid and three-hybrid analyses.
Expression patterns of genes can be probed through the use of microarrays.
Genes can be inactivated or altered with the CRISPR/Cas9 system, as a method of elucidating or manipulating gene function.
UNANSWERED QUESTIONS
As powerful as biotechnology has become, there are still limitations to the reach of molecular biology.
Can personalized genomics become commonplace? The DNA sequencing methods described in this chapter are making the personal genome a reality, at a reasonable cost. And that cost is still declining rapidly. Besides the technological developments, much thought is required about what we will do with all this DNA sequence information when we get it. Ethical considerations must play a role in these decisions.
How do we apply biotechnology to routinely and safely alter human genomes? The CRISPR/Cas9 system is the most recent and robust addition to the toolbox that molecular biologists can use to alter genomes. Safe and effective application to human genomes may become a reality. With more efficient methods, the treatment of human genetic disease by gene therapy may become practical.
Can we find out where proteins function in a cell, and what they do? There is much room for improvement in the methods based on fusion proteins, now used to track the location and function of proteins in cells. Too often, the protein tags used for this work have unintended effects on the activity of the protein to which the tag is attached. The GFP protein works well in many cases, but smaller and brighter tags would be very helpful, along with new ways to link them to proteins.
HOW WE KNOW: New Enzymes Take Molecular Biologists from Cloning to Genetically Modified Organisms
Cohen, S.N., A.C.Y. Chang, H.W. Boyer, and R.B. Helling. 1973. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA 70:3240–3244.
Jackson, D.A., R.H. Symons, and P. Berg. 1972. Biochemical method for inserting new genetic information into DNA of simian virus 40: Circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 69:2904–2909.
DNA cloning is commonplace now, but the idea of combining DNA from two different species created quite a stir in the early 1970s. A convergence of research advances in many laboratories gave rise to a technological revolution.
The first experiment to join two DNA molecules together used a rather laborious strategy. Peter Lobban, a graduate student in Dale Kaiser’s lab at Stanford, first used the enzyme terminal transferase to add “tails” of A residues to one DNA fragment and T residues to another. The poly(A) and poly(T) tails annealed, and the fragments could be covalently joined with DNA ligase (Figure 1). This experiment linked two segments of DNA derived from a bacterial virus, P-22. Paul Berg’s lab soon used the same strategy to link two DNA segments from different species, one from the bacterial virus lambda (λ) and the other from the simian virus SV40. The Berg paper was published in 1972.
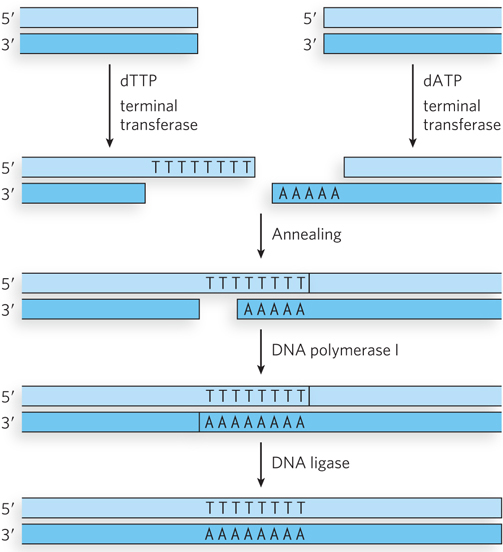
FIGURE 1 A homopolymer tail was added to each DNA segment with the aid of the enzyme terminal transferase. A poly(A) tail was added to one segment, a poly(T) tail to the other. The two complementary tails were annealed. Gaps were filled with the aid of DNA polymerase, and the DNA was joined by DNA ligase.
Meanwhile, Stanley Cohen, also at Stanford, began to study DNA plasmids that made certain disease-causing bacteria resistant to antibiotics (a problem then, as it remains today). His lab soon developed methods for isolating the plasmids and reintroducing them into other bacteria. To get at the genes causing the antibiotic resistance and other features of the plasmid, Cohen wanted to take the plasmids apart and reassemble them. Rather than use the laborious poly(A)–poly(T) tail approach, Cohen relied on restriction enzymes that had recently been discovered. Herbert Boyer and his colleagues at the University of California, San Francisco, had shown that one of these enzymes, EcoRI, cleaves DNA asymmetrically at a 6 bp palindrome (see Table 7-2). The resulting sticky ends could guide the rejoining of the ends by DNA ligase.
At a November 1972 meeting in Hawaii, Cohen and Boyer began a collaboration that led to DNA cloning. They settled on a plasmid called pSC101 from the Cohen lab, which encoded a gene that conferred resistance to tetracycline. The collaborators found that Boyer’s EcoRI enzyme cut the plasmid only once, and not in a region that affected the sequences needed for either replication or tetracycline resistance. By early 1973, they had demonstrated that DNA segments from any source, also derived from cleavage by EcoRI, could be linked to this plasmid. The plasmid, in turn, could be reintroduced and propagated in bacteria. These advances, reported in the Proceedings of the National Academy of Sciences (USA) in late 1973, gave rise to the first DNA cloning patents and a new company (Genentech). More importantly, they set the stage for the rise of biotechnology.
A Dreamy Night Ride on a California Byway Gives Rise to the Polymerase Chain Reaction
Brock, T.D., and H. Freeze. 1969. Thermus aquaticus gen. n. and sp. n., a nonsporulating extreme thermophile. J. Bacteriol. 98:289–297.
Saiki, R.K., S. Scharf, F. Faloona, K.B. Mullis, G.T. Horn, H.A. Erlich, and N. Arnheim. 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle-cell anemia. Science 230:1350–1354.
Sometimes, large advances in science arise through inspiration. In the spring of 1983, Kary Mullis was an employee of the Cetus Corporation in northern California. Hired in 1979 to synthesize oligonucleotides, Mullis discovered that as oligonucleotide synthesis became increasingly automated, he had more and more time to contemplate other projects. He became interested in methods to detect small sequence differences in human DNA, but initially did not make much progress. The idea for the polymerase chain reaction occurred to Mullis one night, in April 1983, as he drove with a friend up the coast. As he described it, he stopped the car and started drawing—DNA molecules hybridizing and lengthening, a chain reaction in which the products of one cycle became the templates for the next.
The first experiment was carried out a few months later, and the first report of the polymerase chain reaction came out in a 1985 paper in Science describing a new procedure for detecting the hemoglobin mutation that causes sickle-cell anemia. The method was spelled out in more detail in publications appearing over the next two years. In the early trials, the polymerase used was an active fragment of the E. coli DNA polymerase I (see Chapter 11). The heating required to denature the DNA after each PCR cycle inactivated this polymerase, so it had to be added again after each cycle.
A side story shows how basic research can contribute to major advances in surprising ways. About two decades before the development of PCR, microbiologist Thomas Brock (at the University of Wisconsin–Madison) initiated some studies of organisms in the hot springs of Yellowstone National Park (Figure 2). In the fall of 1966, he succeeded in culturing a bacterium from a pool called Mushroom Spring, an organism that grew at higher temperatures than were thought possible for living organisms. The new thermophilic bacterium was subsequently named Thermus aquaticus. The heat stability of the proteins in T. aquaticus became highly important to the development of PCR. The DNA polymerase from this bacterium (Taq polymerase) is stable at very high temperatures and is not inactivated by the heating and cooling cycles that are needed to denature and reanneal the DNA during PCR. The incorporation of Taq polymerase into the PCR protocol in the late 1980s allowed the entire procedure to be automated.

FIGURE 2 Hot springs in Yellowstone National Park, one of which is shown here, were the source of the bacterium Thermus aquaticus.
PCR methods were quickly optimized, and new protocols gradually expanded the possible applications. By the end of the 1980s, the technology had utterly transformed the biological sciences.
HOW WE KNOW: Coelenterates Show Biologists the Light
Chalfie, M., Y. Tu, G. Euskirchen, W.W. Ward, and D.C. Prasher. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802–805.
Heim, R., D.C. Prasher, and R.Y. Tsien. 1994. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 91:12501–12504.
Shimomura, O., F.H. Johnson, and Y. Saiga. 1962. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell. Comp. Physiol. 59:223–239.
In 1960, shortly after joining the faculty at Princeton University, Osamu Shimomura began to study the bioluminescence produced by the jellyfish Aequorea victoria. Traveling regularly to the state of Washington to secure specimens, he would take the photo-organs from 20 or 30 jellyfish and squeeze them through rayon gauze. His “squeezate” was slightly luminescent, and he began to purify the molecules responsible. In 1962, his lab reported the purification of a protein associated with what was later named green fluorescent protein, which they called aequorin. The first reference to GFP appears in a footnote in that paper, in which Shimomura described isolating from squeezates a protein that formed solutions that looked greenish in sunlight and yellowish under tungsten lights, and showed very bright, greenish fluorescence in UV light. His subsequent studies gradually showed that GFP had a special property: it contained all the chemistry needed to emit fluorescence on its own. Up to that time, most other proteins known to produce bioluminescence, such as firefly luciferase, required the addition of other molecules to do so.
Douglas Prasher, at the Woods Hole Oceanographic Institution, was the first to appreciate the potential of fusing GFP to another protein and using its fluorescence as a cellular marker for that protein. He succeeded in cloning the gene for GFP and determining its sequence, reporting this advance in 1992. Martin Chalfie, at Columbia University, had also seen the potential of GFP. Collaborating with Prasher, he expressed GFP in E. coli, reporting the results of the work in Science in 1994 (Figure 3). This work realized the potential of the system, showing that GFP expressed in a host organism produced fluorescence without the need for any other protein or factor from the jellyfish. Sergey Lukyanov, in Moscow, showed that variants of GFP could be cloned from the nonbioluminescent Anthozoa of coral reefs, and he managed to expand the color range of this protein class by cloning a red fluorescent protein.

FIGURE 3 The bacteria on the right side of this agar plate are glowing as a result of the expression of green fluorescent protein.
Much of what we now know about the chemistry and general utility of GFP and other fluorescent proteins has resulted from the subsequent work of Roger Tsien, at the University of California, San Diego. His laboratory has constructed many mutants of GFP that produce the range of colors seen in Figure 7-22b. Many of these mutants also improve on the stability and brightness of the fluorophores in the proteins. Since the mid-1990s, GFP and its variants have become a staple of molecular and cellular biology, illuminating the mechanisms and pathways of countless other cellular proteins and processes.