9.3 THE ENZYMES THAT PROMOTE DNA COMPACTION
DNA supercoiling—
Topoisomerases Catalyze Changes in the Linking Number of DNA
All cells, from bacteria to eukaryotes, have enzymes with the sole function of underwinding and relaxing DNA. Topoisomerases increase or decrease the extent of DNA underwinding by changing the linking number. They play an especially important role in the complex changes in DNA topology during replication and DNA packaging.
As we show in later chapters, topoisomerases are crucial to every aspect of DNA metabolism. As a consequence, they are important drug targets for the treatment of bacterial infections and cancer (Highlight 9-2). Inactivating mutations in genes that encode key cellular topoisomerases, the enzymes responsible for the degree of supercoiling in cells, often result in severe growth deficiencies or cell death.
There are two classes of topoisomerases (Table 9-4). Type I topoisomerases break one of the two DNA strands, pass the unbroken strand through the break, and ligate the broken ends; they change Lk in increments of 1. Type II topoisomerases break both DNA strands and change Lk in increments of 2. The DNA is never released from the enzyme during these topological transactions, so uncontrolled relaxation of the DNA does not occur.

312
HIGHLIGHT 9-2 MEDICINE: Curing Disease by Inhibiting Topoisomerases
The topological state of cellular DNA is intimately connected with its function. Without topoisomerases, cells cannot replicate or package their DNA, or express their genes—
Two classes of bacterial topoisomerase inhibitors have been developed as antibiotics. The coumarins, including novobiocin and coumermycin A1, are natural products derived from Streptomyces species. They inhibit the ATP binding of the bacterial type II topoisomerases, DNA gyrase and topoisomerase IV. These antibiotics are not used to treat infections in humans, but research continues to identify clinically effective variants.
The quinolone antibiotics, also inhibitors of bacterial DNA gyrase and topoisomerase IV, first appeared in 1962 with the introduction of nalidixic acid (Figure 1). This compound had limited effectiveness and is no longer used clinically in the United States, but the continued development of this class of drugs eventually led to the introduction of the fluoroquinolones, exemplified by ciprofloxacin (Cipro). The quinolones act by blocking the last step of the topoisomerase reaction in bacteria, the resealing of the DNA strand breaks. Ciprofloxacin is a broad-

Some of the most important chemotherapeutic agents used in cancer treatment are inhibitors of human topoisomerases. Tumor cells generally contain elevated levels of topoisomerases, and agents targeted to these enzymes are much more toxic to the tumors than to most other tissue types. Inhibitors of both type I and type II topoisomerases have been developed as anticancer drugs.
Camptothecin, isolated from a Chinese ornamental tree and first tested clinically in the 1970s, is an inhibitor of eukaryotic type I topoisomerases. Clinical trials indicated limited effectiveness in treating cancer, however, despite its early promise in preclinical work on mice. Two effective derivatives were developed in the 1990s: irinotecan (Campto) and topotecan (Hycamtin), used to treat colorectal cancer and ovarian cancer, respectively (Figure 2). Additional derivatives are likely to be approved for clinical use in the coming years. All of these drugs act by trapping the topoisomerase-
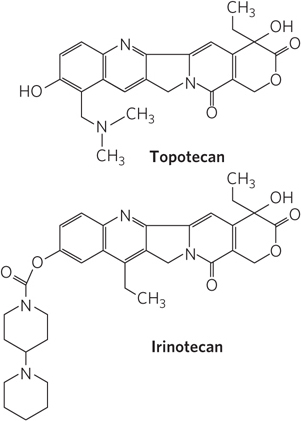
The human type II topoisomerases are targeted by a variety of antitumor drugs, including doxorubicin (Adriamycin), etoposide (Etopophos), and ellipticine (Figure 3). Doxorubicin, effective against several kinds of human tumors, is in clinical use. Most of these drugs stabilize the covalent topoisomerase–

All of these anticancer agents generally increase the levels of DNA damage in targeted, rapidly growing tumor cells, but noncancerous tissues can also be affected, leading to a more general toxicity and unpleasant side effects that must be managed during therapy. As cancer therapies become more effective and survival statistics for cancer patients improve, the independent appearance of new tumors is becoming a greater problem. In the continuing search for new cancer therapies, topoisomerases are likely to remain prominent targets.
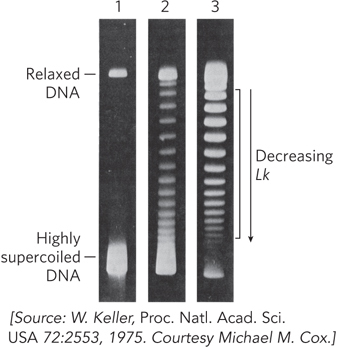
The activity of these enzymes can be observed with agarose gel electrophoresis, which separates DNA species according to their topoisomeric form (Figure 9-18). A population of identical plasmid DNAs with the same linking number migrates as a discrete band during electrophoresis. DNA topoisomers that are more supercoiled are more compact and migrate faster in the gel. Topoisomers with Lk values differing by as little as 1 can be separated by this method, so the changes in linking number induced by topoisomerases are readily detected.
E. coli has at least four individual topoisomerases, I through IV. Topoisomerases I and III are of type I, and they generally relax DNA by introducing transient single-

313
The topoisomerase must both cleave a DNA strand and religate it after the topological change is complete. The phosphodiester bond is not simply hydrolyzed, because this would entail loss of a high-
The Two Bacterial Type II Topoisomerases Have Distinct Functions
Bacterial topoisomerase II, also known as DNA gyrase, can introduce negative supercoils (decrease Lk). This enzyme cleaves both strands of a DNA molecule (thus is a type II topoisomerase) and passes another duplex through the break (see the How We Know section at the end of this chapter). The introduction of negative supercoils alone would put additional strain on the DNA molecule, but gyrase has an additional activity that uses the energy of ATP to drive key conformational changes that counteract the thermodynamically unfavorable introduction of negative supercoils. Bacterial DNA gyrases are the only topoisomerases known to actively introduce negative supercoils.
314
315
Gyrase is composed of two types of subunits, GyrA and GyrB, functioning as a GyrA2GyrB2 heterotetramer (Figure 9-20a). GyrB interacts with DNA and ATP and catalyzes ATP binding and hydrolysis. Parts of GyrB form the entry point for DNA, called the N-

316
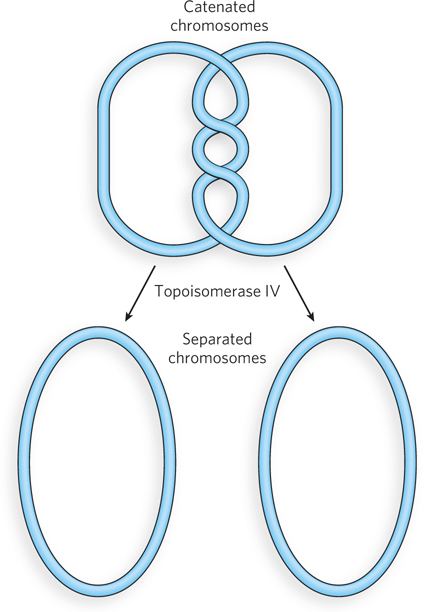
The second bacterial type II topoisomerase, DNA topoisomerase IV, has a very specialized function. Immediately following replication, the circular daughter chromosomes of bacteria are topologically intertwined. Circular DNAs that are intertwined in this way are called catenanes (Figure 9-21). DNA topoisomerase IV unlinks the catenated daughter chromosomes, allowing their proper segregation at cell division. Unlike DNA gyrase, this enzyme does not use ATP and does not introduce negative supercoils.
Eukaryotic Topoisomerases Have Specialized Functions in DNA Metabolism
Eukaryotic cells also have type I and type II topoisomerases. The type I enzymes are called topoisomerases I and III. They function primarily in relieving tension and resolving topological problems in DNA during replication and repair. The type II enzymes are topoisomerases IIα and IIβ (see Table 9-4). Eukaryotic type II topoisomerases cannot underwind DNA (introduce negative supercoils), but they can relax both positive and negative supercoils. They function in all aspects of eukaryotic DNA metabolism, resolving a range of topological problems that occur during replication, transcription, and repair. They play an especially important role in the condensation of chromosomes into highly structured chromatin.
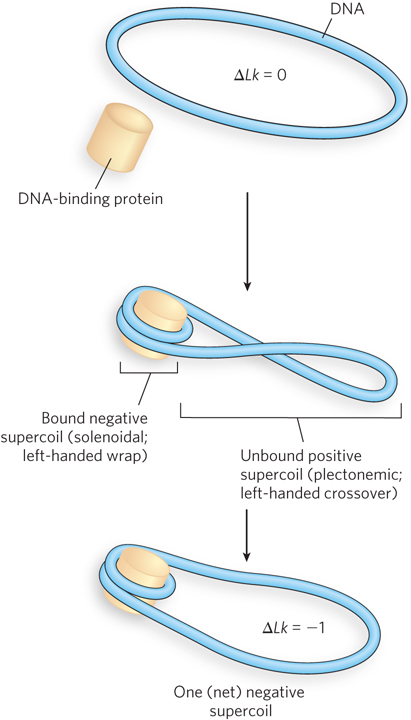
Although eukaryotes do not have an enzyme that can introduce negative supercoils into DNA, when a circular DNA is isolated from a eukaryotic cell (e.g., a plasmid from yeast), it is negatively supercoiled. This reflects the generally underwound state of cellular DNA in eukaryotic cells. One probable origin of negative supercoils in eukaryotic DNA is the tight wrapping of the DNA around a nucleosome in chromatin, which introduces a negative solenoidal supercoil without changing the number of turns in the molecule (see Chapter 10). Because the Lk remains unchanged, the negative solenoidal supercoil has to be compensated for by a positive supercoil elsewhere in the DNA (Figure 9-22). The type II topoisomerases relax the unbound positive supercoils that arise in this way. The bound and stabilized negative supercoils are left behind, conferring a net negative superhelicity on the DNA. Next to the histones that make up the nucleosomes, type II topoisomerases are the most abundant proteins in chromatin.
317
Figure 9-23 shows the reaction catalyzed by eukaryotic type II topoisomerases. The multisubunit enzyme binds a DNA molecule (step 1). The gated cavities above and below the bound DNA are the N-

SMC Proteins Facilitate the Condensation of Chromatin
Whereas topoisomerases influence supercoiling by changing the linking number of chromosomes, SMC proteins (structural maintenance of chromosomes) promote chromosome condensation by creating physical contact between segments of DNA that may otherwise be quite distant from each other in the chromosome, or even on different chromosomes. This class of protein is found in all organisms. In eukaryotes, where their function is best understood, these enzymes have integral roles in DNA condensation and chromosome segregation during mitosis, as well as in DNA repair. They perform their tasks by lining up along the DNA and binding to each other, providing a link between distant parts of the chromosome.
SMC proteins have five distinct domains (Figure 9-24a). The amino-

All bacteria have at least one SMC protein that functions as a homodimer to assist in compacting the genome, whereas eukaryotes generally have six SMC proteins that work in defined pairs as heterodimers with different functions (Figure 9-24c). Electron microscopy reveals the flexible V shape of these proteins (Figure 9-24d). The SMC1-
318
The SMC1-
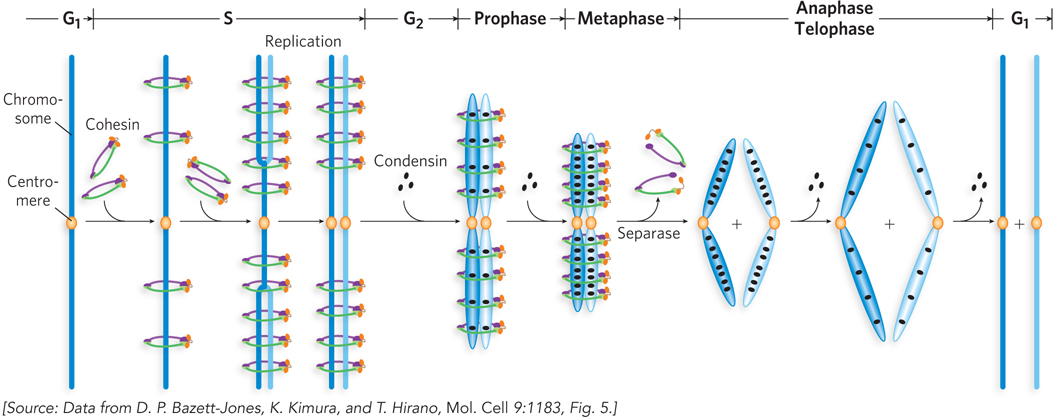
Interaction among the head groups of multiple SMC proteins has the potential to produce several different architectures, such as rings, rosettes, and filaments (Figure 9-26). It is not yet clear whether the ringed cohesin tethers around sister chromatids are intra-

The SMC2-
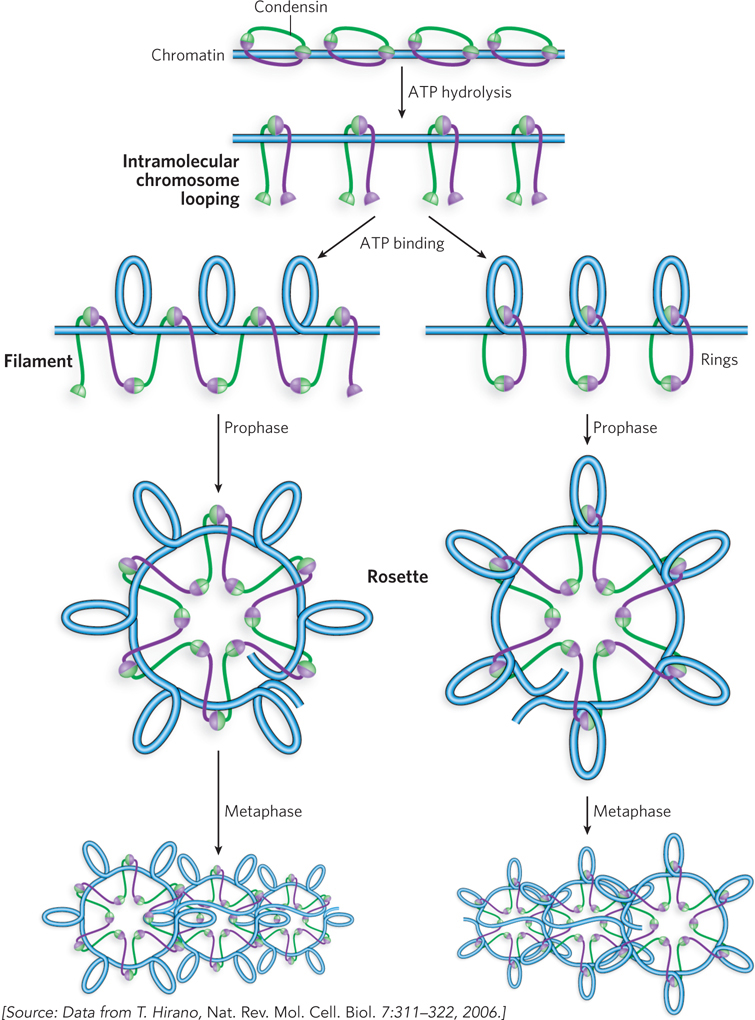
319
The topoisomerases and SMC proteins enable cells to deal with the complex topological changes occurring as DNA strands separate during replication, repair, and transcription, as well as the extraordinary degree of DNA compaction required in every cell. The compaction is maintained by additional specialized DNA-
SECTION 9.3 SUMMARY
Topoisomerases catalyze the underwinding and relaxation of DNA. On a molecular level, topoisomerases catalyze changes in the linking number.
The two classes of topoisomerases, type I and type II, change Lk in increments of 1 or 2, respectively, per catalytic event.
The reactions catalyzed by DNA topoisomerases involve the formation of transient covalent DNA-
enzyme intermediates, usually in the form of a phosphotyrosyl linkage. Bacterial DNA gyrases introduce negative supercoils.
Topoisomerases have functions specific to DNA metabolism, such as unlinking catenated bacterial DNA after replication or relaxing supercoils formed by unwinding during replication and transcription.
320
Condensation of cellular chromosomes is facilitated by SMC proteins, including cohesins and condensins. Cohesins tether the sister chromatid products of DNA replication, and condensins provide a general structural scaffold for chromosome condensation.
UNANSWERED QUESTIONS
In large chromosomes, with their DNA highly complexed with proteins and thereby constrained, topological challenges accompany every process in DNA metabolism. The simple movement of a DNA polymerase through the DNA during replication (defining a structure called a replication fork) leads to overwinding ahead of the fork and underwinding behind it. Some of the challenges are extraordinary, such as when two replication forks converge in a eukaryotic chromosome, or when intertwined chromosomal DNAs must be separated at cell division. A complete understanding of DNA metabolism in cells will require more detailed information on how every system interfaces with the proteins that solve topological problems.
321
What is the role of bacterial SMC proteins? Bacteria generally have at least one SMC protein, and sometimes several. Mutational loss of the major bacterial SMC protein usually leads to defects in the condensation and segregation of chromosomes at cell division. Recent work indicates that this SMC protein is recruited to the daughter replication origins and participates in the mechanism that ensures proper segregation. However, much remains to be learned about this process.
What is the function of the eukaryotic SMC5-
SMC6 proteins? The most enigmatic of the eukaryotic SMC proteins is the SMC5- SMC6 pair. So far, we know that this protein pair functions primarily in processes such as DNA recombination and repair, and much evidence now implicates SMC5- SMC6 in chromosome dynamics during meiosis. However, the precise contribution of this pair of proteins remains unknown. How does topoisomerase III function in DNA metabolism? In both bacteria and eukaryotes, topoisomerase III is closely tied to the function of helicases in the RecQ family. In humans, defects in the genes encoding RecQ family helicases lead to genetic diseases, including Bloom and Werner syndromes, that are characterized by genomic instability and an increased propensity to develop cancer. These enzymes are essential to many aspects of DNA metabolism. For example, the topoisomerase III–
2RecQ pairing in bacteria plays a critical role in resolving topological problems that accompany the convergence of replication forks. Again, much remains to be learned about the mechanics of these complicated transactions.
322
The Discovery of Supercoiled DNA Goes through Twists and Turns
Lebowitz, J. 1990. Through the looking glass: The discovery of supercoiled DNA. Trends Biochem. Sci. 15:202–
Vinograd, J., J. Lebowitz, R. Radloff, R. Watson, and P. Laipis. 1965. The twisted circular form of polyoma viral DNA. Proc. Natl. Acad. Sci. USA 53:1104–
By 1962, the double-
Analytical ultracentrifugation measures the migration of molecules through a density gradient when ultracentrifugal force is applied (see Figure 16-2); molecules that migrate farther through the gradient have a larger sedimentation coefficient (S). Using this and other methods, researchers found three forms of polyoma DNA in the isolated preparations, which migrated with sedimentation coefficients of 20S, 16S, and 14S. Because the 20S and 16S forms (forms I and II, respectively) were most abundant, the researchers focused on those two. The two forms had the same molecular weight, so the different sedimentation velocities had to involve different conformations. Form I could be isolated in almost pure preparations, if care was taken while preparing the DNA.
Both the Dulbecco and the Vinograd groups published reports indicating that form I could be converted to form II by the addition of reagents that promote DNA strand cleavage. The researchers developed a model in which they assumed form I to be circular and form II to be linear. The observed kinetics suggested that the conversion occurred in a single step. This challenged the model, because two strands would have to be cleaved to generate the linear molecule, and both would have to be broken at the same position.
The Vinograd group decided to examine all three forms of polyoma DNA (20S, 16S, and 14S) by electron microscopy. Philip Laipis carried out the first experiments. His examination of forms I and II yielded the unexpected result that both were circular (Figure 1). Laipis was a relatively inexperienced undergraduate, and some of the other researchers in the lab initially assumed he had made a mistake in preparing the samples. However, several repetitions yielded the same result. Only form III (14S) was linear. Careful controls eliminated any possibility that the result reflected a selective elimination of linear forms during preparation of the DNA for examination; when the researchers premixed measured amounts of forms III and II and then examined them, the linear and circular DNAs were always there in the expected proportions. Additional kinetic studies showed that only one strand break was needed to convert form I to form II. Cleaving form I with endonuclease I (which cleaves both strands) produced only the form III (14S) DNA, with no form II. Forms I and II also had identical buoyant densities, making it unlikely that some non-

The important clue lay in the electron micrographs. The form I molecules appeared much more twisted on themselves than the form II molecules. Denaturation experiments showed that the strands of form II could be separated, but those of form I could not. The researchers gradually established that form II was a nicked DNA circle. Understanding form I required a little more work. A comment from colleague Robert Sinsheimer led Vinograd to focus on the twisted nature of the form I DNA. His subsequent modeling with phone cords was documented in 1990 in a delightful retrospective article by Jacob Lebowitz, one of the authors on the 1965 paper. Although the term “supercoiling” was not yet in common use, the discovery of the supercoiled nature of polyoma DNA opened an entirely new field of investigation.
323
The First DNA Topoisomerase Unravels Some Mysteries
Wang, J.C. 1971. Interaction between DNA and an Escherichia coli protein ω. J. Mol. Biol. 55:523–
Over the course of the twentieth century, life scientists were getting used to a fundamental idea: if a change occurs in any cellular structure, there is at least one enzyme that catalyzes it. The first discovery of an enzyme involved in DNA supercoiling was made by James Wang and reported in 1971.
Working at the University of California, Berkeley, Wang had initiated studies of superhelicity in small DNAs that could be isolated from bacteria. Focusing on the DNA from bacteriophage λ, he noticed that E. coli extracts contained a macromolecule that converted the superhelical form of the circular DNA to a relaxed form. He did what any good molecular biologist would do: he purified the macromolecule. The result was a protein that he dubbed the ω (omega) protein. Later, it became known as DNA topoisomerase I.
Initially, the purification was incomplete. However, Wang could establish that the macromolecule was a protein, because its activity was not lost after extended dialysis (using a membrane that allowed small molecules to escape but retained larger proteins); activity was lost when the preparation was heated to 50°C (a temperature that denatures most proteins). Wang demonstrated that the conversion of a DNA circle with 150 superhelical turns to a relaxed circle did not happen in one step. Using sedimentation velocity studies and electron microscopy, he showed that the change was progressive, with one or a few superhelical turns lost in each catalytic step. The activity affected only negative, not positive, superhelicity.
Wang concluded that the enzyme had two activities: a nicking activity and a strand-

324
DNA Gyrase Passes the Strand Test
Brown, P.O., and N.R. Cozzarelli. 1979. A sign inversion mechanism for enzymatic supercoiling of DNA. Science 206: 1081–

In 1976, Martin Gellert and colleagues reported the discovery of a second topoisomerase in E. coli. The enzyme, DNA gyrase, had the novel property that it could introduce negative supercoils into DNA, hydrolyzing ATP in the process. DNA gyrase was quickly shown to be critical to DNA replication and other processes, and there was great interest in determining how it worked. Many researchers expected that DNA gyrase generated a net negative superhelicity by relaxing positive supercoils, using a mechanism much like that exhibited by the ω protein, with the creation of a break in one strand and rotation of that strand about the other.
Nicholas Cozzarelli and colleagues, at the University of Chicago, began to focus on experimental observations that did not fit this scheme. First, when active DNA gyrase was acting on a DNA, and the gyrase-
Pooling this and other information, Cozzarelli proposed a very different mechanism for gyrase action, one he dubbed “sign inversion” (Figure 3). He imagined that in a circular DNA, gyrase would bind to two segments of DNA that crossed over each other, thereby stabilizing a positive crossover, or node. The creation of such a node would necessarily create a compensating negative node elsewhere in the DNA molecule. Gyrase would then invert the sign of the bound node by breaking both DNA strands, passing the unbroken DNA segment through the break, and resealing the break on the other side. This would change the sign of the node to negative and effectively fix two negative supercoils in the DNA.

The sign inversion model made a novel and unique prediction. DNA gyrase would do something very different from the ω protein: it would change superhelicity in increments of 2 rather than increments of 1. This prediction was not trivial to test. Supercoiled circular DNA (such as plasmid DNA) is isolated from cells as a heterogeneous mixture of topoisomers with a roughly Gaussian distribution of linking numbers. Gyrase could shift the center of that distribution, but highlighting individual reaction steps to observe the predicted Lk increments of 2 would be difficult. Cozzarelli and his student, Patrick O. Brown, found a way to overcome the problem.
325
They initially focused on a particularly small circular DNA, a plasmid of about 2,400 bp called p15. Such a small DNA limited the total number of topoisomers in the Gaussian distribution and facilitated the separation of one topoisomer from another on an agarose gel. Using the ω protein, Brown and Cozzarelli took a naturally supercoiled preparation of p15 DNA and relaxed it completely. They then ran the DNA on an agarose gel under conditions in which the topoisomers of p15 were well separated. They cut the most abundant topoisomer out of the gel and extracted it, effectively isolating a DNA preparation with one topoisomer only.
With a topologically pure DNA in hand, the key experiment could be done. The researchers added enough DNA gyrase (about two heterotetramers per DNA molecule) to ensure that essentially every DNA circle had a bound gyrase. After incubating the DNA and gyrase for 3 minutes, they added ATP, but only enough (30 μM, or about one-
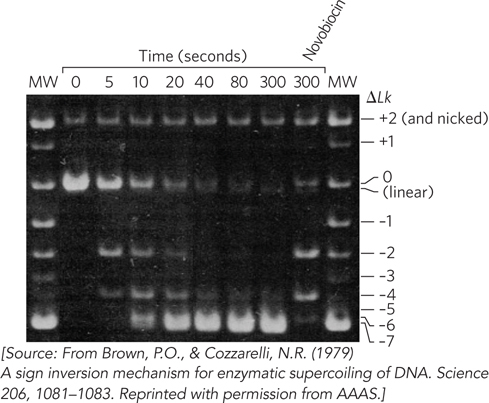
Brown and Cozzarelli carried out one additional test. After 5 minutes, the p15 DNA was highly supercoiled. They then added novobiocin, an antibiotic that inhibits supercoiling but not relaxation by gyrase (see Highlight 9-2). After another 30 minutes of incubation, much of the DNA had been substantially relaxed (see Figure 4). The topoisomers present included species with superhelicity changes of 0, –2, and –4 relative to the starting material. This demonstrated that gyrase promoted both supercoiling and relaxation of DNA in increments of 2. The result fulfilled a key expectation of any enzymatic reaction—
These advances helped explain the mechanism of action of a range of important antibiotics and antitumor drugs (see Highlight 9-2). They were among a string of important contributions from the Cozzarelli lab, first at Chicago and later at the University of California, Berkeley. With an ebullient personality and a creative intellect, Cozzarelli inspired a generation of scientists, both as a mentor and a colleague. Cozzarelli died due to complications of a treatment for Burkitt’s lymphoma in 2006, at the age of 67. But his lab motto “Blast Ahead” lives on.
326