13.4 NONHOMOLOGOUS END JOINING
Recombination allows an accurate restoration of broken chromosomes, a considerable virtue given the importance of maintaining genomic integrity. However, recombination is complicated and requires the action of dozens of proteins. Sometimes DSBs occur when recombinational DNA repair is not feasible, such as during phases of the cell cycle when no sister chromatids are present. At these times, another path is needed to avoid the cell death that would result from a broken chromosome. That alternative is provided by nonhomologous end joining (NHEJ). The broken chromosome ends are simply processed and ligated back together.
Nonhomologous End Joining Repairs Double-Strand Breaks
Nonhomologous end joining is an important pathway for DSBR in all eukaryotes, and it has also been detected in some bacteria. In general, the importance of NHEJ increases with genomic complexity. Only a few bacteria seem to have NHEJ systems. In yeast, most DSBs are repaired by recombination, and only a few by NHEJ. In mammals, many DSBs occurring outside meiosis are repaired by NHEJ. These patterns reflect differences in cellular lifestyles. In all eukaryotic cells, recombinational DNA repair is the preferred DSBR pathway during the S and G2 phases of the cell cycle, when chromosomes are being replicated and paired prior to cell division. Finding a homolog to direct the repair process by recombination is readily accomplished at these times. NHEJ is critical to the repair of DSBs that arise during the G1 and the static G0 phases of the cell cycle, when homologous chromosomes are not readily aligned. Differentiated mammalian cells may divide rarely, if at all, and typically spend much more time in the G1 and G0 phases than do yeast cells, which may divide every few hours. When DSBs occur during these phases, the enzymes that promote NHEJ are rapidly activated.
476
Unlike homologous recombinational repair, NHEJ does not conserve the original DNA sequence. When a DSB occurs during G1 or G0, a protein complex forms at each broken end of the chromosome, and the two DNA-
NHEJ is a mutagenic process, and a smaller genome, such as that of yeast, has relatively little tolerance for the loss of information. The small genomic alterations may be tolerable in mammalian somatic cells, however, because they are not in the germ line and will not be inherited, and they are balanced by the undamaged information on the homolog in each diploid cell. Indeed, the propensity of NHEJ to create mutations has led to its being recruited in somatic cells as a source of variation in the production of genes encoding antibodies (see Chapter 14).
Nonhomologous End Joining Is Promoted by a Set of Conserved Enzymes
In eukaryotes, at least nine proteins are used in the multiple steps of NHEJ (Table 13-2). The reaction is initiated at a DSB by the binding of a heterodimer consisting of the proteins Ku70 and Ku80 (“KU” are the initials of the individual with scleroderma whose serum autoantibodies were used to identify this protein complex; the numbers refer to the approximate molecular weights of the subunits). The Ku proteins are conserved in almost all eukaryotes. Both subunits of the Ku70-
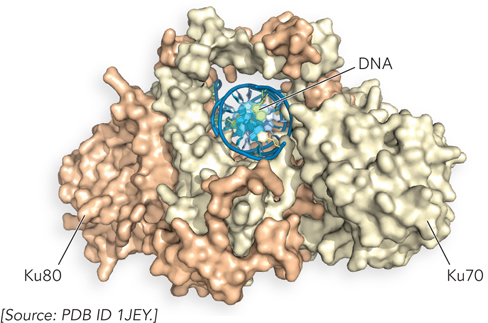
NHEJ proceeds in three major stages (Figure 13-26). In the first stage, Ku70-
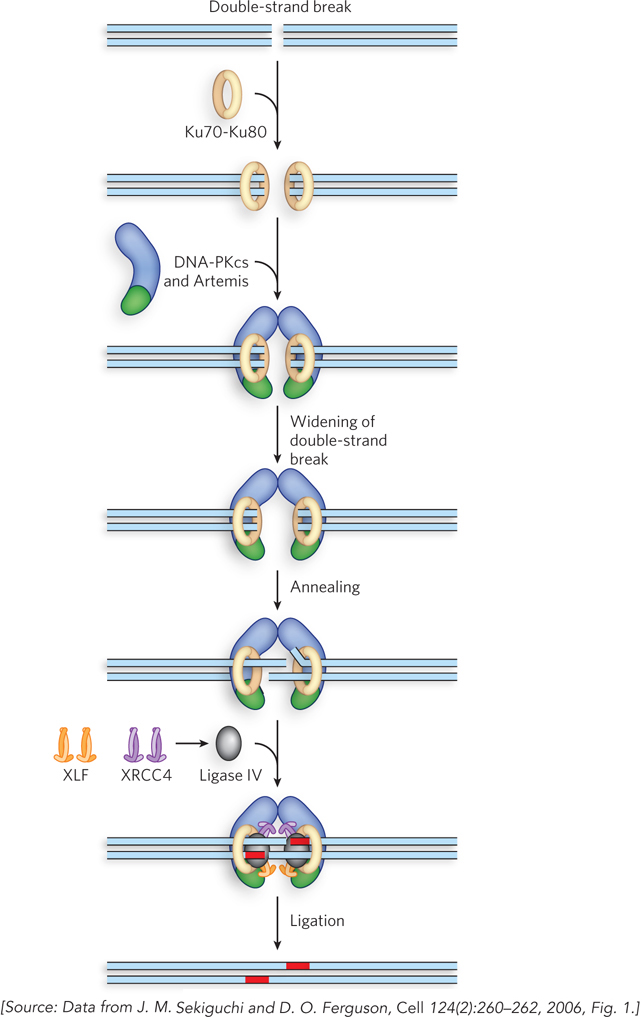
477
DNA ends are usually not joined randomly by NHEJ. Instead, when a DSB occurs, the ends are generally constrained by the structure of chromatin and thus remain close together; they are rarely linked to the ends of other chromosomes, because all eukaryotic chromosome ends are protected by telomeres. Very rare events linking end sequences that are normally far apart in the chromosome, or on different chromosomes, may be responsible for occasional dramatic and usually deleterious genomic rearrangements.
Recombination Systems Are Being Harnessed for Genome Editing
As should be clear by now, recombination events are initiated with double-
478
These goals will be met slowly, as problems and obvious ethical issues abound. The programmable nucleases have significant tendencies to cleave DNA at sites that are off-
SECTION 13.4 SUMMARY
Nonhomologous end joining is critical to the repair of double-
strand breaks that arise during the G1 and static G0 phases of the eukaryotic cell cycle. NHEJ is more important in cells that spend a greater amount of time in G1 and G0, such as the somatic cells of more complex eukaryotes. NHEJ is promoted by the well-
conserved Ku70- Ku80 protein complex, along with additional complexes containing nuclease, polymerase, and ligation activities. Double-
strand break repair makes a critical contribution to genome editing methods that utilize programmable nucleases to generate targeted double- strand breaks.
UNANSWERED QUESTIONS
In every area of molecular biology, there is a need to reconcile biochemistry with the observations made in living cells, using genetics and functional genomics. Recombination systems and processes are complex, and accurately reconstituting them in vitro from purified components remains a major challenge. Here are some of the significant questions in the field.
How often do replication forks collapse? It is not yet clear how often DNA template lesions halt replication forks in a manner requiring replication restart, and how often lesions are simply bypassed by the replication machinery. Some in vitro studies show a potential for bypass even for lesions in the leading strand. However, in vivo studies show that replication ceases for a period when sufficient DNA damage is introduced into the genome. Although we understand the outlines of the major recovery pathways, there are doubtless many variants yet to be elucidated—
along with some undiscovered enzymes— that respond to different classes of lesions and the many different DNA structures found at stalled forks. In a eukaryotic chromosome, what parameters determine where crossovers occur during meiotic recombination? Much remains to be discovered about the structural features of chromosomes that define crossover hot spots and how numbers of crossovers are controlled.
What factors remain to be discovered in double-
strand break repair, and how do they work? In studies of recombinational DNA repair, many new proteins are still being discovered. Some of the newer discoveries involve protein factors that regulate recombination, or link it to replication checkpoints or other aspects of chromosome structure or cell division. The complexities are illustrated by proteins such as Spo11. The double- strand cleavage reaction of Spo11 has not yet been replicated in vitro, perhaps due to a requirement for other protein factors not yet purified (or not yet discovered). After Spo11 has cleaved a chromosome, additional enzymes must degrade the 5′-ending strand to create the 3′ single- stranded extensions needed for strand invasion. The identity of the nuclease that processes these DNA ends is not yet clear. A complete DSBR reaction has yet to be reconstituted in vitro. How is recombination coordinated with other aspects of DNA metabolism? Regulation is an increasingly visible theme in this field of research. Recombination must be directed at locations where it is needed, and prevented elsewhere. When a replication fork stalls, recombinational repair systems must arrive quickly and address the situation at hand. The intricate coordination required to keep these processes on track is critical to genomic integrity, and even survival, and understanding it will keep many molecular biology laboratories engaged for decades to come.
What is the tension-
sensing mechanism that facilitates proper chromosomal segregation in eukaryotic cell division? Some of the participating proteins have been discovered, but much remains to be done in this area of molecular biology.
479
A Motivated Graduate Student Inspires the Discovery of Recombination Genes in Bacteria
Clark, A.J. 1996. RecA mutants of E. coli K12: A personal turning point. Bioessays 18:767–
Clark, A.J., and A.D. Margulies. 1965. Isolation and characterization of recombination deficient mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 53:451–
Sometimes it is the student who challenges the professor. This is what happened in 1962 at the University of California, Berkeley, when first-

The two researchers decided to use bacterial conjugation as a way to measure recombination events. As Joshua Lederberg and E. L. Tatum had demonstrated in 1946, some bacteria harbor plasmids that can be transferred between cells. These F plasmids sometimes integrate themselves into the bacterial chromosome, creating strains (Hfr strains) that can convey parts of their chromosome to other cells at high frequency. When DNA is transferred, alleles from the donor DNA can be transmitted to the recipient’s chromosome by recombination. Margulies and Clark used replica plating, a technique devised by Esther Lederberg and Joshua Lederberg in 1952, to search for mutants.

They used two strains of E. coli. The chosen Hfr donor strain could not grow unless leucine was included in the growth medium (this strain was denoted leu−). The recipient strain, lacking an F plasmid, had a mutation leading to a requirement for adenine (ade−). Conjugational crosses between the two strains produced recombinants that could grow in the absence of both leucine and adenine (leu+ade+).
The recipient strain was treated with the mutagen 1-
Using a piece of sterile velvet, Margulies replicated the pattern of colonies on the master plate onto the third plate. The transferred cells underwent conjugational mating with cells in the lawn of donor Hfr bacteria. Successful conjugation and recombination produced high-
The procedure was laborious, but Margulies, working under Clark’s guidance, persevered. After months of careful controls and screening more than 2,000 mutagenized recipient strains, Margulies found two strains that had a recombination defect. Later work established that these strains had mutations in what became known as the recA gene. Clark and Margulies published their results in the Proceedings of the National Academy of Sciences in 1965; their paper has been cited countless times. The work launched John Clark into a productive career in elucidating recombination mechanisms. Sadly, Ann Dee Margulies, the intrepid graduate student, died of cancer in 1980, at the age of 40.
480
A Biochemical Masterpiece Catches a Recombination Protein in the Act
Keeney, S., C.N. Giroux, and N. Kleckner. 1997. Meiosis-
Following the proposal of the double-
The answer was delivered in a biochemical exercise marked by both determination and elegance. The trick was to isolate the protein bound to the cleaved 5′ ends of the DSB, but this was no simple task. Every meiotic cell has scores of such cleavage events, and they are spaced along chromosomes containing millions of base pairs, bound by hundreds of different proteins.
The researchers’ first step was one that biochemists often use: amplification of the signal. Keeney, Kleckner, and others had found that when steps subsequent to formation of the DSB, such as the rapid degradation of the 5′-ending strands, were blocked, covalent protein-
Using rad50S cells as an enriched source of the protein-
In the second step, the researchers separated protein-
The procedure was carried out in parallel on rad50S cells and on cells with a mutation that prevents DSB formation (as control). Doing so on a large, preparative scale yielded the results shown in Figure 1. Two bands, with apparent molecular weights of 34,000 and 45,000, were seen in the rad50S samples but not in the control samples. The two proteins were excised from the gel and identified by tandem mass spectrometry as a contaminant and as Spo11, respectively. More controls were carried out to solidify the case that Spo11 is the protein bound to the break sites. In one particularly compelling experiment, Spo11 was immunoprecipitated from rad50S cells with covalently linked DNA fragments from a known recombination hot spot.
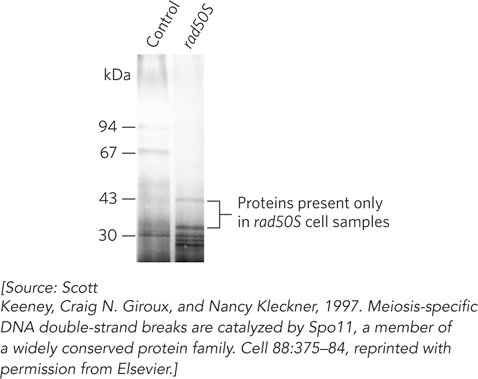
The Spo11-
481