17.4 EXCEPTIONS PROVING THE RULES
Initial studies of the genetic code suggested that it was universal and without variation. And, as we’ve seen, this universality implies that life evolved from a common ancestor. Presumably, once the code had developed in LUCA, it became locked in place because descendants could not tolerate changes in the genetic code. For example, a change in a codon that specifies lysine to one that specifies leucine would result in a Leu residue replacing every Lys residue in every protein in the cell. Clearly, such a global change would be fatal to the cell, so it is unlikely that such codon changes could occur, even over a long span of evolutionary time. How the translation machinery evolved in LUCA remains one of the greatest questions in evolutionary biology, and with no “missing link” cells, we may never know the answer. But even though we do not know how the process of translation evolved, it is interesting to contemplate the evolutionary hurdles that must have been overcome to arrive at this process.
We now know that there are some exceptions to the rules of the genetic code. It is not entirely universal after all. Does this mean that the ancestral cell did not perfect the code before modern cells diverged from it? The evidence suggests not. Most of the exceptions support the evolution of a common code in LUCA from which a few changes, in some circumstances, evolved.
Evolution of the Translation Machinery Is a Mystery
With the discovery of ribozymes, the hypothesis of the RNA world became a very plausible model for the beginning of life. In the RNA world, RNA catalyzes essentially all the chemical reactions needed for life, and there are many examples of catalytic RNAs in cells today—
Any hypotheses about how translation evolved must account for each part of the translation machinery. At least 20 tRNAs, 20 aminoacyl-
605
Finally, how did the genetic code evolve? Was it a random act of evolution, or did the amino acids somehow participate in generation of the code as we know it today? Did all 20 tRNAs appear individually, or did one appear through random mutation and then diversify into the rest? Did the first code use triplet sequences, or was it simpler, using dinucleotide sequences to code for fewer amino acids? How was a reading frame established? And how did the point of termination develop?
These are some of the challenges to hypothesizing how the translation process evolved. One hypothesis is presented in Highlight 17-1. All cells have the complete translation machinery; there are no cells with “missing links” to tell us the story more directly. However, certain scientific approaches can help illuminate how the process evolved. For example, as noted above, there are exceptions to the genetic code, and we can examine them for insights about its evolution. We also know that RNAs can perform many enzymatic reactions, supporting the RNA world hypothesis. Ongoing research is defining the minimal genetic and protein requirements for a living cell, which will identify the essential genetic and protein requirements for life—
HIGHLIGHT 17-1 EVOLUTION: The Translation Machinery
How the ribosome, tRNAs, and the genetic code evolved is one of the most perplexing and fascinating areas of evolutionary history. In the RNA world, nucleic acids not only stored genetic information but also performed all the catalytic reactions necessary for life. In modern cells, most enzymes are proteins, which are more efficient catalysts than RNAs. The leap from nucleic acid to protein requires very complicated machinery, and it presumably evolved in steps. Furthermore, LUCA could not predict that proteins would be superior to RNAs as catalysts, so we can presume that the first proteins were made to serve some other purpose. What were the evolutionary steps leading to the translation apparatus, and how did natural selection produce them? As an intellectual exercise, let’s consider just one of several possible explanations.
First of all, why would a cell want a protein when it is already using RNA to catalyze cellular metabolism? Harry Noller suggests that the first proteins evolved to help RNA ribozymes fold properly. For example, the high negative charge of the nucleic acid backbone hinders the close approach of nucleic acid helices, but the charge can be mitigated by a protein’s basic amino acid side chains. Indeed, most ribozymes in modern cells (including ribosomes) contain a protein component, and many ribosomal proteins are at junctions of RNA helices and may stabilize their proximity. Therein lies a possible selection pressure for an RNA-
What about the building blocks of proteins? Several amino acids were most likely present in the primordial soup, but how were they linked together in a way that is coded by an RNA without aminoacyl-
Now we have the rudiments of a simpler RNA world–
The early translation process may have operated at very low fidelity, yet it served the purpose of making structural peptides that enhanced RNA catalytic function. For example, early tRNAs may not have been entirely selective for one amino acid, but may have selected for certain amino acid properties such as charge, polarity, or hydrophobicity. This level of accuracy might be acceptable if the functions of early proteins were purely structural. But even for a structural role, the pressures of natural selection would eventually result in high fidelity in the translation process. Although cells did not “know” that amino acid side chains offered much greater chemical potential than nucleic acids for both structure and catalysis, they had developed a mechanism to code for protein. And selection could do the rest: the evolution of the protein world would proceed according to the rules of natural selection. Proteins that are better catalysts than their RNA counterparts would eventually take over much of the job of catalytic RNAs, and aminoacyl-
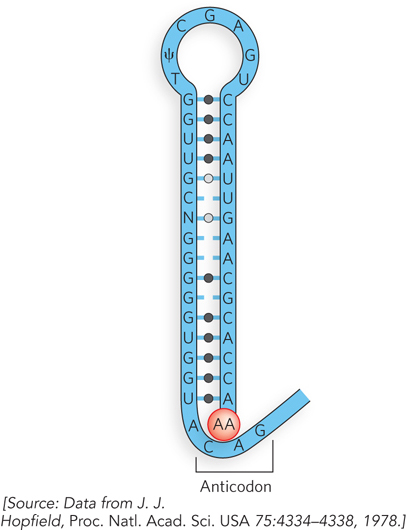
The evolution of the genetic code is another area of lively debate. Proposals range from a code that simply arose at random and became frozen in time, to one in which the amino acids themselves participated directly in code development. For the latter proposal, consider the hypothetical tRNA structure in Figure 1. The anticodon would participate in selecting the correct amino acid, possibly through favorable contacts between the amino acid and a particular triplet nucleotide sequence, depending on the chemical nature of the amino acid. Indeed, experimental support for this hypothesis exists. The preferential affinity of amino acids for certain nucleic acid sequences has been studied, and it is reported that some amino acids exhibit preferential binding to the anticodons of the tRNAs that encode them. It’s also possible that not all 20 amino acids were present early on, and that only a two-
Mitochondrial tRNAs Deviate from the Universal Genetic Code
Phylogeny tells us that the exceptions to the genetic code are derived from a single, universal code, because the exceptions are rare and occur in different branches of the tree of life (see Chapter 8). But in what situations could changes to the genetic code be viable? Even one change would have a global impact on cellular function, because every protein in the cell is made according to the same set of coding instructions. In other words, a single change in a codon–
With this in mind, we can make a few hypotheses about the types of deviations from the genetic code that might be plausible. For example, we can propose that the most easily altered codons are termination codons, which are not located in the middle of genes. If a stop codon were recruited to code for an amino acid by altering the anticodon of a tRNA, that codon could be placed in internal positions of certain genes as the organism evolved (another stop codon could be used for termination). In this way, a particular amino acid would be inserted in the middle of the gene where the stop codon is placed. We can also hypothesize that evolution of this type of exception to the code would have a higher probability in an organism of low genetic complexity (i.e., only a few genes) than in an organism of high complexity, because fewer proteins would be affected. These two hypotheses are largely confirmed.
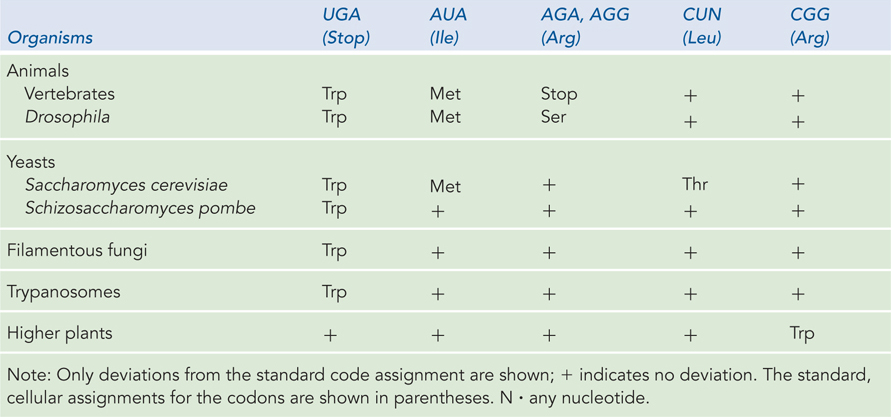
Mitochondria are a prime example of how the genetic code can be altered. Mitochondria are thought to be the descendants of early bacterial cells that were engulfed by eukaryotic cells and proved beneficial because of their unique capacity to perform aerobic metabolism, thereby conferring this advantageous capability on early anaerobic eukaryotes. Over time, this symbiotic relationship relieved the mitochondrial genome of most of its genes, transferring them to the nucleus of the host cell. But mitochondria retained a small genome of their own, with a limited set of genes. Researchers noticed the first deviations from the genetic code when sequencing mitochondrial DNA (mtDNA). A fascinating aspect of the mitochondrial genome is that it encodes a unique set of tRNAs, just for use in decoding the mtDNA. This feature of mitochondria permits changes in their tRNAs without interfering with the information flow of the cellular genome. As predicted for alterations evolving from the standard code, the most common codon changes in mitochondria involve stop codons.
Genetic code changes in mitochondria are essentially the result of an exquisitely streamlined flow of genetic information. Vertebrate mtDNAs encode 13 proteins, 2 rRNAs, and 22 tRNAs. Instead of the minimum of 32 tRNAs needed for the standard, cellular code, the 22 mitochondrial tRNAs can decipher all possible codons by slight alterations in the rules of the code. For example, only one tRNA is used for each of four codon families. In each case, a single tRNA recognizes the four different codons, each with the same first two nucleotides. Each of these mitochondrial tRNAs has a U in the first (wobble) position of the anticodon (that base-
606
If all mitochondrial tRNAs recognize more than one codon, yet another deviation from the rules of the standard genetic code can be inferred. Normally, tryptophan and methionine are specified by one codon each. In mitochondria, the tRNA specifying tryptophan recognizes the UGG codon, but it also recognizes UGA, which is a termination codon in the standard code. The AUG codon for methionine is used in mitochondria to initiate translation, but the standard isoleucine codon, AUA, specifies methionine at internal positions. In the mitochondria of mammals, codons AGA and AGG, which usually specify arginine, are termination codons. These same mitochondrial codons in the fruit fly specify serine. Examples of the known coding variations in mitochondria are summarized in Table 17-4.
The low complexity of the mitochondrial genome has allowed continued evolution, which has resulted in streamlining of the genetic code. However, there are also a few examples in which the code has been altered in free-
607
608
Initiation and Termination Rules Have Exceptions
Changes in the code need not be absolute; a codon might not always encode the same amino acid. In most organisms we find some examples of amino acids being inserted at positions that are not specified in the standard code. Two examples are the occasional use of GUG (valine) or UUG (leucine) as an initiation codon. This occurs only for those genes in which the GUG or UUG codon is properly located in the mRNA (see Chapter 18).
Another example is the insertion of selenocysteine (Sec)—sometimes referred to as the twenty-
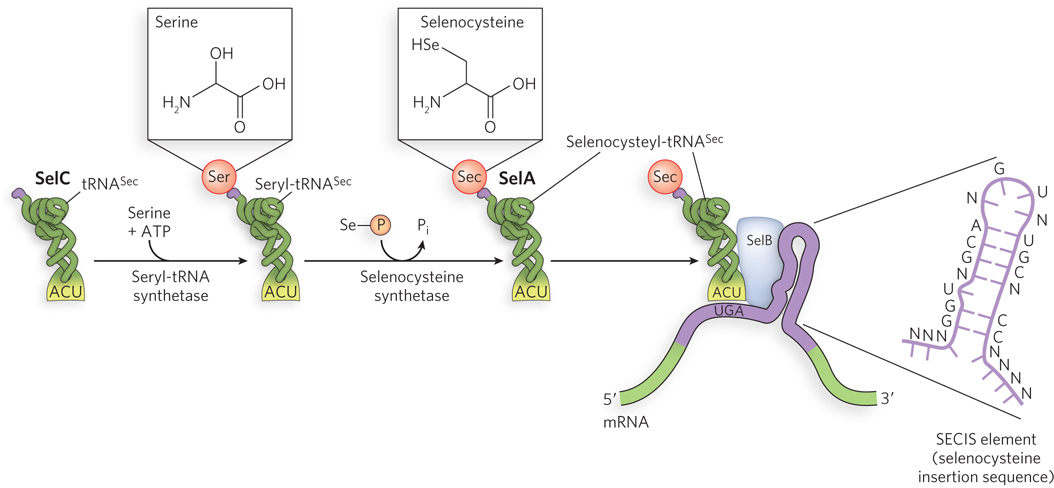
This process of incorporation of Sec residues has been extensively studied in E. coli. Of the gene products involved, SelC is a tRNA (tRNASec) that accepts serine initially. SelA is an enzyme that uses selenophosphate to convert the serine to selenocysteine, forming Sec-
The evolution of genetic code changes in small genomes such as those of mitochondria, the use of a few alternative initiation codons, and the use of a termination codon to incorporate selenocysteine—
609
When several codons encode the same amino acid and require multiple tRNAs, not all of the codons are used with equal frequency. In a phenomenon called codon bias, some codons for a particular amino acid are used more frequently (sometimes much more frequently) than others. The tRNAs for the frequently used codons are often present at much higher concentrations than the tRNAs for the rarely used codons. For example, there are six codons for leucine (see Figure 17-4). In bacteria, CUG is used often to encode Leu. However, in fungi closely related to Candida, CUG is used only rarely as a Leu codon and is often entirely absent in highly expressed proteins. A change in the coding sense of CUG would thus have a much smaller effect on fungal cell metabolism than might be expected if all Leu codons were used equally.
The coding change for the Leu codons may have occurred by a gradual loss of CUG codons in genes and of the tRNA that recognizes CUG as a Leu codon, followed by a capture event—
SECTION 17.4 SUMMARY
The genetic code, tRNAs, and translation must have evolved piecemeal, without protein components, and this evolution must have had a selective advantage, even at its earliest stages. Several hypotheses exist, but evidence to support any of them is limited.
The genetic code is largely universal. Most exceptions occur in mitochondrial DNA, a small genome genetically isolated from the nucleus and relatively free to undergo evolutionary code changes; many of these changes involve altered stop codons, yielding a streamlined genetic code that requires only 22 tRNAs.
The few examples of genetic code alterations outside mtDNA usually involve the conversion of termination codons, in keeping with a common ancestry for the genetic code from which all variants are derived.
UNANSWERED QUESTIONS
The genetic code has been deciphered and is of paramount importance to virtually every investigation in molecular biology. However, important fundamental questions remain. The nature of wobble pairing and the influence of tRNA structure on codon-
How do nucleotides outside the anticodon influence the structure of tRNA for wobble pairing? The use of noncanonical base pairs explains how wobble pairing can happen. But nucleotide substitutions outside the anticodon also affect wobble pairing in some way, perhaps through conformational changes when tRNA binds the ribosome.
Why do tRNAs have so many modified bases? The proportion of modified bases in tRNAs can approach 20%, and many genes are devoted to synthesizing these modified bases. Yet we still know very little about the functions of these many modifications.
Did the three classes of RNA—
tRNA, mRNA, and rRNA— coevolve, as we presume? This seems like a gigantic leap. If they did not coevolve, what might the individual functions have been for each class, leading them to funnel into one central process? How did the protein components that work with RNAs evolve? What functions did the earliest proteins serve? Did early proteins simply serve structural roles to bind and stabilize catalytic RNA, only to take over the catalytic roles later?
How did the translation machinery evolve? It is nothing short of mind-
boggling to imagine how the translation machinery evolved in the first place. One challenge is the huge number of factors required for translation. The whole process could not have evolved all at once. What were the individual steps, and what forces of natural selection were at work?
610
Transfer RNA Connects mRNA and Protein
Hoagland, M.B., M.L. Stephenson, J.F. Scott, L.I. Hecht, and P.C. Zamecnik. 1958. A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem. 231:241–
Once Francis Crick had hypothesized the existence of an adaptor molecule to bridge the information gap between mRNA and protein, the race was on to find it. It may seem obvious now, but before the discovery of aminoacyl-
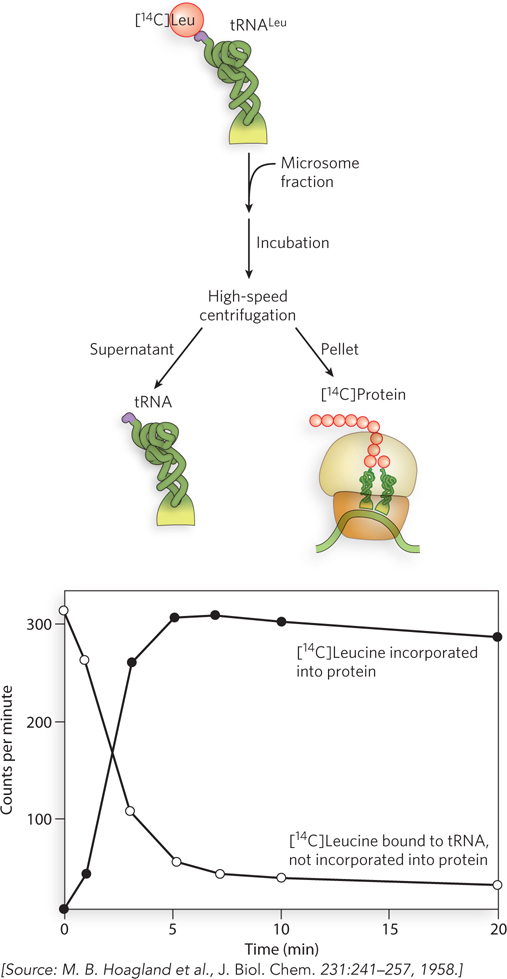
The race to find the adaptor was won by Mahlon Hoagland and Paul Zamecnik. They prepared a cell extract that contained the soluble tRNA and the enzymes needed to charge tRNA with amino acids. When [14C]leucine was added to the extract, the radiolabeled amino acid became attached to its tRNA. To show that this really was the adaptor molecule, Hoagland and Zamecnik demonstrated that the [14C]leucyl-

611
Proteins Are Synthesized from the N-Terminus to the C-Terminus
Dintzis, H.M. 1961. Assembly of the peptide chains of hemoglobin. Proc. Natl. Acad. Sci. USA 47:247–
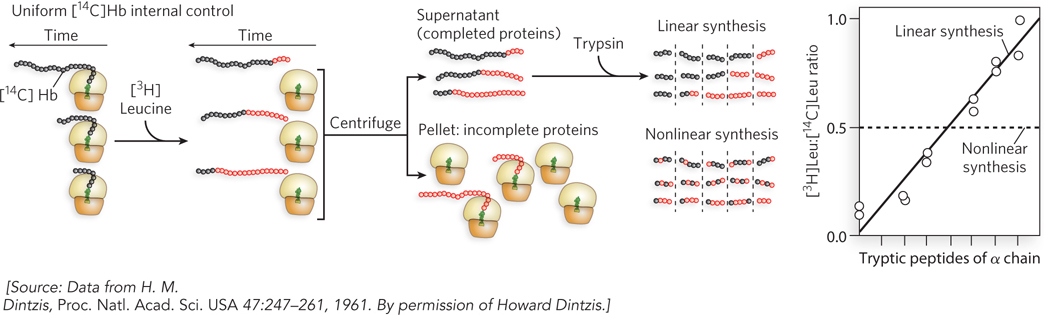
After the adaptor tRNA was discovered, the basic flow of information among the three biopolymers, DNA → RNA → protein, was understood. However, the order in which amino acids were connected to form a protein polymer was still unknown. To solve this, Howard Dintzis designed an ingenious experiment. His idea was to feed [3H]leucine to living cells and follow protein synthesis over time. Leucine was known to be one of the most abundant amino acids in proteins, and he assumed it would be located in random places from one end of a polypeptide to the other. Would incorporation of [3H]leucine start at one end of a newly synthesized protein, or would the labeled amino acid appear uniformly throughout the length of the new protein at all time points? This might sound like an easy experiment, but there were big technical hurdles. First, Dinztis had to follow only one protein, yet a cell makes many proteins all at once. Second, protein sequencing had not yet been invented, but Dintzis had to find some way to follow the sequence in which leucine was incorporated in the protein chain.
The beauty in Dintzis’s experimental design lies in how he circumvented these difficulties. He knew that immature red blood cells (reticulocytes) turn off the synthesis of most proteins except hemoglobin, which is composed of two chains, α and β. Furthermore, he could use trypsin (a protease) to digest the α chain and could separate the fragments by paper electrophoresis, although he couldn’t determine the ordering of the fragments along the protein. So Dintzis added [3H]leucine to reticulocytes, lysed cells at various times, separated the α and β chains, and analyzed the α chain by tryptic digestion and electrophoresis. Full-
If protein chains are not made in a defined order, all peptides should have a similar ratio of 3H to 14C (Figure 2). But if proteins are made in a linear order, peptides will vary in their 3H:14C ratio, and the part of the protein made last should contain a higher 3H:14C ratio than the parts made earlier. The results were unambiguous and striking! The peptides differed greatly in 3H:14C ratio, ruling out a random order of synthesis. Further, the tryptic peptides could be ordered to form a gradient of radioactivity. Hence, it was apparent that hemoglobin is synthesized from one end to the other. To determine the direction of synthesis, Dintzis digested the α chain with carboxypeptidase, which specifically removes amino acids from the C-
612
The Genetic Code In Vivo Matches the Genetic Code In Vitro
Terzaghi, E., Y. Okada, G. Streisinger, J. Emrich, M. Inouye, and A. Tsugita. 1966. Change of a sequence of amino acids in phage T4 lysozyme by acridine-
The experiments that defined the genetic code were brilliant, and this was Nobel Prize–
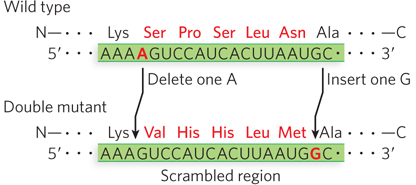
The investigators used a protease to digest lysozyme, subjected the fragments to electrophoresis, and studied the resulting peptide maps. Comparing maps of the double-
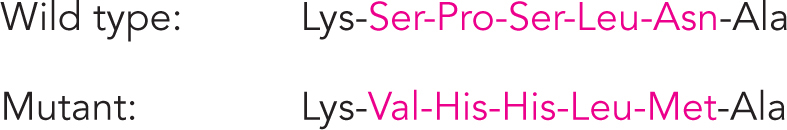
This result supported the triplet reading frame with no internal punctuation marks, as described by Crick. It could also be used to solve whether the genetic code in living cells is the same as that determined in extracts, by comparing the wild-
613