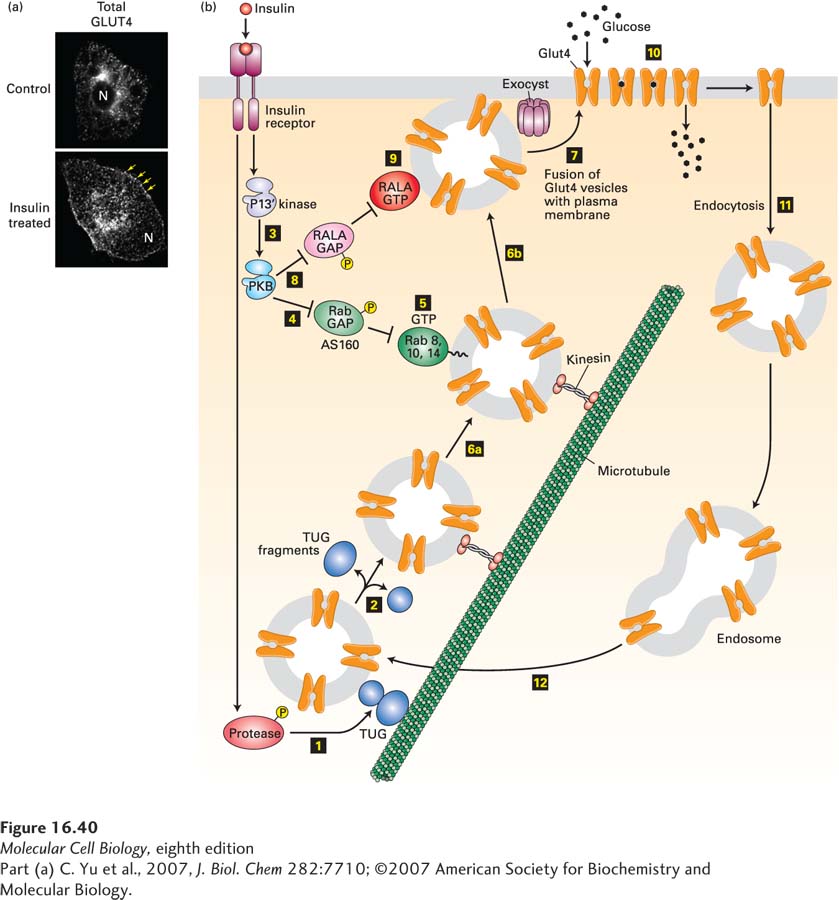
In Fat and Muscle Cells, Insulin Triggers Fusion of Intracellular Vesicles Containing the GLUT4 Glucose Transporter to the Plasma Membrane
The released insulin circulates in the blood and binds to insulin receptors, which are present on many different kinds of cells, including muscle and adipocyte cells. The insulin receptor, a receptor tyrosine kinase, activates several signal transduction pathways, including the one leading to the activation of protein kinase B (PKB; see Figure 16-29). In this case, the main action of the PKB signaling pathway—an increase in uptake of glucose from the blood—is manifest within minutes. Since glucose uptake is the rate-limiting step in glucose utilization, this action results in rapid lowering of the blood glucose level.
Like those of most body cells, the plasma membranes of fat and muscle cells contain the GLUT1 glucose transporter, which allows the cell to import sufficient glucose for its basal metabolic needs. Fat and muscle cells also express large amounts of the insulin-responsive glucose transporter GLUT4; in resting (unstimulated) cells, virtually all of the GLUT4 is localized to small vesicles in the cytosol (Figure 16-40). While some GLUT4 is in endosomes, most is in a unique small organelle termed the GLUT4 storage vesicle (GSV). In these vesicles, much of the GLUT4 is tethered to the Golgi matrix, a network of coiled-coil proteins surrounding the Golgi complex, by a protein termed TUG.
EXPERIMENTAL FIGURE 16-40Insulin stimulation of fat cells induces translocation of GLUT4 from intracellular vesicles to the plasma membrane. (a) Cultured adipose cells engineered to express a chimeric protein comprising GLUT4 with a green fluorescent protein (GFP) fused to its C-terminus were visualized with a confocal fluorescence microscope. In the absence of insulin, virtually all of the GLUT4 is in intracellular membranes. Treatment with insulin triggers fusion of the GLUT4-containing membranes with the plasma membrane. Arrows highlight GLUT4 present at the plasma membrane; N indicates the position of the nucleus. (b) In fat and muscle cells, insulin signaling acts in multiple steps to increase the level of GLUT4 at the plasma membrane. In resting cells, the majority of the GLUT4 protein is localized to specialized GLUT4 storage vesicles (GSVs), tethered to Golgi matrix proteins by the TUG protein. Binding of insulin to the insulin receptor leads to activation of a protease (step 1) that cleaves the TUG protein, releasing GLUT4-containing vesicles (step 2), which then move along microtubules, powered by a kinesin motor (see Chapter 18), to the cell surface. Insulin also activates PKB (step 3; see Figure 16-29). PKB then phosphorylates the Rab GAP protein AS160 (step 4), inhibiting its ability to accelerate GTP hydrolysis by Rab8, Rab10, and Rab14. These Rab proteins accumulate in their active GTP-bound states (step 5) and allow the GLUT4 storage vesicles to move along microtubules to the cell surface (steps 6a and 6b). Finally, these GSVs fuse with the plasma membrane (step 7). This step is catalyzed by the exocyst and also by another monomeric GTP-binding protein, RALA. PKB stimulates this membrane fusion event by phosphorylating and thus inactivating the RALA GAP protein RGC (step 8), allowing RALA to accumulate in its active GTP-bound state (step 9). The resultant increase in plasma membrane GLUT4 allows the cell to incorporate glucose from the extracellular fluids at a rate about 10 times that of unstimulated cells (step 10). Following removal of insulin, the plasma membrane GLUT4 is internalized by endocytosis (step 11) and eventually transported to GSVs (step 12). Many other proteins, not shown here, participate in these signaling and vesicle budding and fusion events. See J. Bogan, 2012, Annu. Rev. Biochem.81:507, D. Leto and A. Saltiel, 2012, Nat. Rev. Mol. Cell Biol.13:383, and J. Belman et al., 2014, Rev. Endocr. Metab. Disord.15:55.
In addition to activating the PI-3 kinase/PKB pathway, the insulin receptor phosphorylates several other target proteins. Together, these signals cause the movement of GLUT4-containing vesicles to the plasma membrane and then the fusion of these vesicles with the plasma membrane. The resulting immediate tenfold increase in the number of GLUT4 molecules on the cell surface increases glucose influx proportionally, thus lowering blood glucose (Figure 16-40b):
Insulin triggers, by a signaling pathway that is only now being completely identified, activation of a protease that catalyzes a site-specific endoproteolytic cleavage of TUG, separating the N-terminal GLUT4-binding segment from the rest of the protein that is anchored to the Golgi matrix. This cleavage allows the GLUT4 vesicles to move to the plasma membrane.
Recall that certain monomeric GTP-binding proteins are essential for the budding of intracellular transport vesicles (e.g., Sar proteins; see Figures 14-6 and 14-8); others, the Rabs, are essential for vesicle fusion (see Figure 14-10). PKB phosphorylates, and by so doing inactivates, two GAP proteins termed AS160 and RGC. In the basal unstimulated state, these GAPs inhibit Rab function by enhancing their rates of GTP hydrolysis, thus keeping the GLUT4 storage vesicles from moving to and fusing with the plasma membrane. Inhibition of these GAPs allows these monomeric GTP-binding proteins in fat and muscle cells to accumulate in their active GTP-bound state. These proteins catalyze multiple steps in the GLUT4 pathway, including transport of the GLUT4 storage vesicles along microtubules to the cell surface and, together with the exocyst (see Chapter 14), fusion of these vesicles with the plasma membrane (see Figure 16-40b).
As the blood glucose level drops, insulin secretion and insulin blood levels drop, and insulin receptors are no longer activated as strongly. In fat and muscle cells, plasma-membrane GLUT4 becomes internalized by endocytosis and stored in intracellular membranes, lowering the level of cell-surface GLUT4 and thus of glucose import.