2.6The Amino Acid Sequence of a Protein Determines Its Three-Dimensional Structure
The Amino Acid Sequence of a Protein Determines Its Three-Dimensional Structure
How is the elaborate three-
Agents such as urea or guanidinium chloride effectively disrupt a protein’s noncovalent bonds. Although the mechanism of action of these agents is not fully understood, computer simulations suggest that they replace water as the molecule solvating the protein and are then able to disrupt the van der Waals interactions stabilizing the protein structure. The disulfide bonds can be cleaved reversibly by reducing them with a reagent such as β-mercaptoethanol (Figure 2.52). In the presence of a large excess of β-mercaptoethanol, the disulfides (cystines) are fully converted into sulfhydryls (cysteines).
50
Most polypeptide chains devoid of cross-
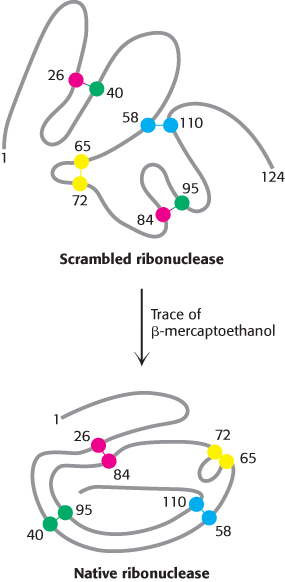
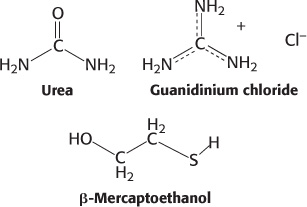
Anfinsen then made the critical observation that the denatured ribonuclease, freed of urea and β-mercaptoethanol by dialysis (Section 3.1), slowly regained enzymatic activity. He perceived the significance of this chance finding: the sulfhydryl groups of the denatured enzyme became oxidized by air, and the enzyme spontaneously refolded into a catalytically active form. Detailed studies then showed that nearly all the original enzymatic activity was regained if the sulfhydryl groups were oxidized under suitable conditions. All the measured physical and chemical properties of the refolded enzyme were virtually identical with those of the native enzyme. These experiments showed that the information needed to specify the catalytically active structure of ribonuclease is contained in its amino acid sequence. Subsequent studies have established the generality of this central principle of biochemistry: sequence specifies conformation. The dependence of conformation on sequence is especially significant because of the intimate connection between conformation and function.
A quite different result was obtained when reduced ribonuclease was reoxidized while it was still in 8 M urea and the preparation was then dialyzed to remove the urea. Ribonuclease reoxidized in this way had only 1% of the enzymatic activity of the native protein. Why were the outcomes so different when reduced ribonuclease was reoxidized in the presence and absence of urea? The reason is that the wrong disulfides formed pairs in urea. There are 105 different ways of pairing eight cysteine molecules to form four disulfides; only one of these combinations is enzymatically active. The 104 wrong pairings have been picturesquely termed “scrambled” ribonuclease. Anfinsen found that scrambled ribonuclease spontaneously converted into fully active, native ribonuclease when trace amounts of β-mercaptoethanol were added to an aqueous solution of the protein (Figure 2.54). The added β-mercaptoethanol catalyzed the rearrangement of disulfide pairings until the native structure was regained in about 10 hours. This process was driven by the decrease in free energy as the scrambled conformations were converted into the stable, native conformation of the enzyme. The native disulfide pairings of ribonuclease thus contribute to the stabilization of the thermodynamically preferred structure.
Similar refolding experiments have been performed on many other proteins. In many cases, the native structure can be generated under suitable conditions. For other proteins, however, refolding does not proceed efficiently. In these cases, the unfolded protein molecules usually become tangled up with one another to form aggregates. Inside cells, proteins called chaperones block such undesirable interactions. Additionally, it is now evident that some proteins do not assume a defined structure until they interact with molecular partners, as we will see shortly.
51
Amino acids have different propensities for forming α helices, β sheets, and turns
How does the amino acid sequence of a protein specify its three-
|
Amino acid |
α helix |
β sheet |
Reverse turn |
|---|---|---|---|
|
Glu |
1.59 |
0.52 |
1.01 |
|
Ala |
1.41 |
0.72 |
0.82 |
|
Leu |
1.34 |
1.22 |
0.57 |
|
Met |
1.30 |
1.14 |
0.52 |
|
Gln |
1.27 |
0.98 |
0.84 |
|
Lys |
1.23 |
0.69 |
1.07 |
|
Arg |
1.21 |
0.84 |
0.90 |
|
His |
1.05 |
0.80 |
0.81 |
|
Val |
0.90 |
1.87 |
0.41 |
|
Ile |
1.09 |
1.67 |
0.47 |
|
Tyr |
0.74 |
1.45 |
0.76 |
|
Cys |
0.66 |
1.40 |
0.54 |
|
Trp |
1.02 |
1.35 |
0.65 |
|
Phe |
1.16 |
1.33 |
0.59 |
|
Thr |
0.76 |
1.17 |
0.96 |
|
Gly |
0.43 |
0.58 |
1.77 |
|
Asn |
0.76 |
0.48 |
1.34 |
|
Pro |
0.34 |
0.31 |
1.32 |
|
Ser |
0.57 |
0.96 |
1.22 |
|
Asp |
0.99 |
0.39 |
1.24 |
|
Note: The amino acids are grouped according to their preference for α helices (top group), β sheets (middle group), or turns (bottom group). Source: T. E. Creighton, Proteins: Structures and Molecular Properties, 2d ed. (W. H. Freeman and Company, 1992), p. 256. |
|||
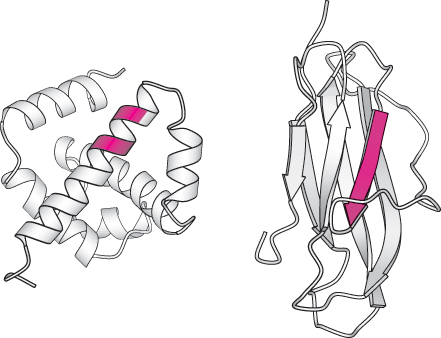
Studies of proteins and synthetic peptides have revealed some reasons for these preferences. Branching at the β-carbon atom, as in valine, threonine, and isoleucine, tends to destabilize α helices because of steric clashes. These residues are readily accommodated in β strands, where their side chains project out of the plane containing the main chain. Serine and asparagine tend to disrupt α helices because their side chains contain hydrogen-
Can we predict the secondary structure of a protein by using this knowledge of the conformational preferences of amino acid residues? Accurate predictions of secondary structure adopted by even a short stretch of residues have proved to be difficult. What stands in the way of more-
52
Protein folding is a highly cooperative process
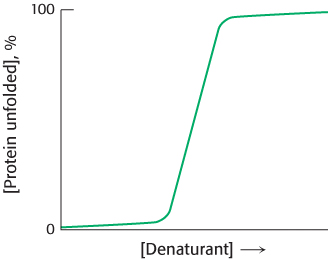
Proteins can be denatured by any treatment that disrupts the weak bonds stabilizing tertiary structure, such as heating, or by chemical denaturants such as urea or guanidinium chloride. For many proteins, a comparison of the degree of unfolding as the concentration of denaturant increases reveals a sharp transition from the folded, or native, form to the unfolded, or denatured form, suggesting that only these two conformational states are present to any significant extent (Figure 2.56). A similar sharp transition is observed if denaturants are removed from unfolded proteins, allowing the proteins to fold.
The sharp transition seen in Figure 2.56 suggests that protein folding and unfolding is an “all or none” process that results from a cooperative transition. For example, suppose that a protein is placed in conditions under which some part of the protein structure is thermodynamically unstable. As this part of the folded structure is disrupted, the interactions between it and the remainder of the protein will be lost. The loss of these interactions, in turn, will destabilize the remainder of the structure. Thus, conditions that lead to the disruption of any part of a protein structure are likely to unravel the protein completely. The structural properties of proteins provide a clear rationale for the cooperative transition.
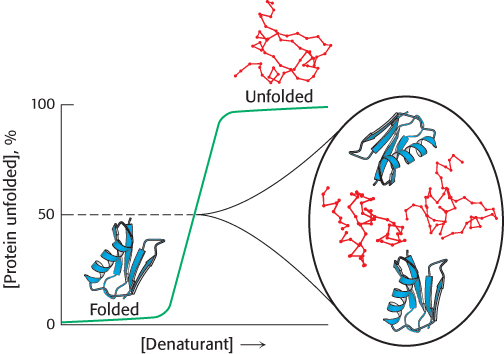
The consequences of cooperative folding can be illustrated by considering the contents of a protein solution under conditions corresponding to the middle of the transition between the folded and the unfolded forms. Under these conditions, the protein is “half folded.” Yet the solution will appear to have no partly folded molecules but, instead, look as if it is a 50/50 mixture of fully folded and fully unfolded molecules (Figure 2.57). Although the protein may appear to behave as if it exists in only two states, this simple two-
53
Proteins fold by progressive stabilization of intermediates rather than by random search

How does a protein make the transition from an unfolded structure to a unique conformation in the native form? One possibility a priori would be that all possible conformations are sampled to find the energetically most favorable one. How long would such a random search take? Consider a small protein with 100 residues. Cyrus Levinthal calculated that, if each residue can assume three different conformations, the total number of structures would be 3100, which is equal to 5 × 1047. If it takes 10−13 s to convert one structure into another, the total search time would be 5 × 1047 × 10−13 s, which is equal to 5 × 1034 s, or 1.6 × 1027 years. In reality, small proteins can fold in less than a second. Clearly, it would take much too long for even a small protein to fold properly by randomly trying out all possible conformations. The enormous difference between calculated and actual folding times is called Levinthal’s paradox. This paradox clearly reveals that proteins do not fold by trying every possible conformation; instead, they must follow at least a partly defined folding pathway consisting of intermediates between the fully denatured protein and its native structure.
The way out of this paradox is to recognize the power of cumulative selection. Richard Dawkins, in The Blind Watchmaker, asked how long it would take a monkey poking randomly at a typewriter to reproduce Hamlet’s remark to Polonius, “Methinks it is like a weasel” (Figure 2.58). An astronomically large number of keystrokes, on the order of 1040, would be required. However, suppose that we preserved each correct character and allowed the monkey to retype only the wrong ones. In this case, only a few thousand keystrokes, on average, would be needed. The crucial difference between these cases is that the first employs a completely random search, whereas, in the second, partly correct intermediates are retained.
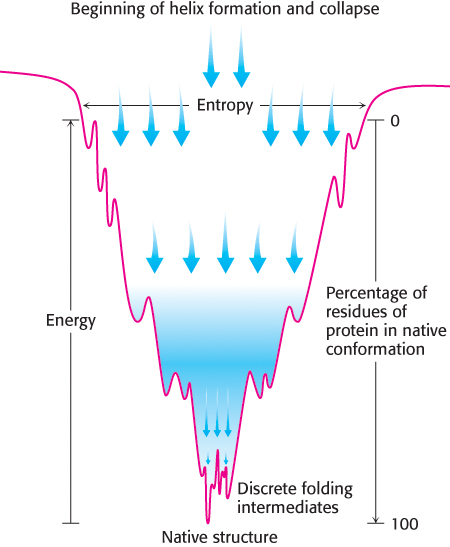
The essence of protein folding is the tendency to retain partly correct intermediates. However, the protein-
A simulation of the folding of a protein, based on the nucleation-
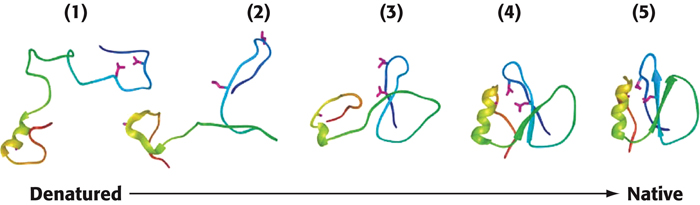
54
Prediction of three-dimensional structure from sequence remains a great challenge
The prediction of three-
Investigators are exploring two fundamentally different approaches to predicting three-
55
Some proteins are inherently unstructured and can exist in multiple conformations
The discussion of protein folding thus far is based on the paradigm that a given protein amino acid sequence will fold into a particular three-
Our first example is a class of proteins referred to as intrinsically unstructured proteins (IUPs). As the name suggests, these proteins, completely or in part, do not have a discrete three-
Another class of proteins that do not adhere to the paradigm is metamorphic proteins. These proteins appear to exist in an ensemble of structures of approximately equal energy that are in equilibrium. Small molecules or other proteins may bind to different members of the ensemble, resulting in various complexes, each having a different biochemical function. An especially clear example of a metamorphic protein is the chemokine lymphotactin. Chemokines are small signaling proteins in the immune system that bind to receptor proteins on the surface of immune-
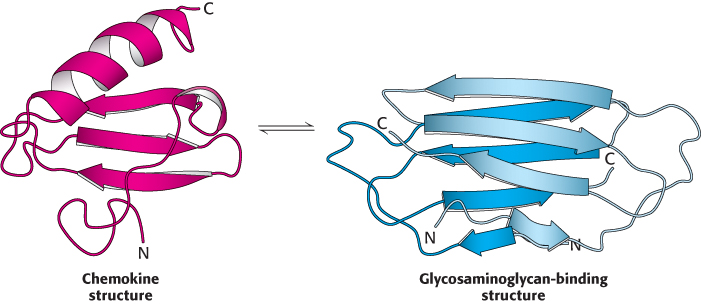
56
Note that IUPs and metamorphic proteins effectively expand the protein-
Protein misfolding and aggregation are associated with some neurological diseases
 Understanding protein folding and misfolding is of more than academic interest. A host of diseases, including Alzheimer disease, Parkinson disease, Huntington disease, and transmissible spongiform encephalopathies (prion disease), are associated with improperly folded proteins. All of these diseases result in the deposition of protein aggregates, called amyloid fibrils or plaques. These diseases are consequently referred to as amyloidoses. A common feature of amyloidoses is that normally soluble proteins are converted into insoluble fibrils rich in β sheets. The correctly folded protein is only marginally more stable than the incorrect form. But the incorrect form aggregates, pulling more correct forms into the incorrect form. We will focus on the transmissible spongiform encephalopathies.
Understanding protein folding and misfolding is of more than academic interest. A host of diseases, including Alzheimer disease, Parkinson disease, Huntington disease, and transmissible spongiform encephalopathies (prion disease), are associated with improperly folded proteins. All of these diseases result in the deposition of protein aggregates, called amyloid fibrils or plaques. These diseases are consequently referred to as amyloidoses. A common feature of amyloidoses is that normally soluble proteins are converted into insoluble fibrils rich in β sheets. The correctly folded protein is only marginally more stable than the incorrect form. But the incorrect form aggregates, pulling more correct forms into the incorrect form. We will focus on the transmissible spongiform encephalopathies.
One of the great surprises in modern medicine was that certain infectious neurological diseases were found to be transmitted by agents that were similar in size to viruses but consisted only of protein. These diseases include bovine spongiform encephalopathy (commonly referred to as mad cow disease) and the analogous diseases in other organisms, including Creutzfeld–
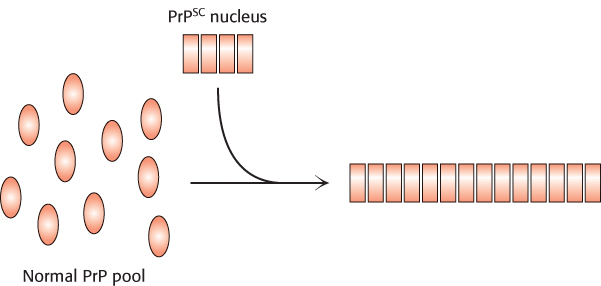
How does the structure of the protein in the aggregated form differ from that of the protein in its normal state in the brain? The normal cellular protein PrP contains extensive regions of α helix and relatively little β strand. The structure of the form of PrP present in infected brains, termed PrPSC, has not yet been determined because of challenges posed by its insoluble and heterogeneous nature. However, a variety of evidence indicates that some parts of the protein that had been in α-helical or turn conformations have been converted into β-strand conformations (Figure 2.62). The β strands of largely planar monomers stack on one another with their side chains tightly interwoven. A side view shows the extensive network of hydrogen bonds between the monomers. These fibrous protein aggregates are often referred to as amyloid forms.
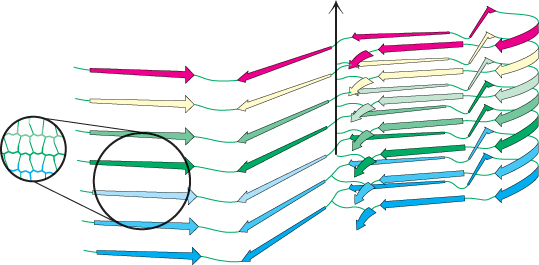
With the realization that the infectious agent in prion diseases is an aggregated form of a protein that is already present in the brain, a model for disease transmission emerges (Figure 2.63). Protein aggregates built of abnormal forms of PrPSC act as sites of nucleation to which other PrP molecules attach. Prion diseases can thus be transferred from one individual organism to another through the transfer of an aggregated nucleus, as likely happened in the mad cow disease outbreak in the United Kingdom that emerged in the late 1980s. Cattle fed on animal feed containing material from diseased cows developed the disease in turn.
57
Amyloid fibers are also seen in the brains of patients with certain noninfectious neurodegenerative disorders such as Alzheimer and Parkinson diseases. For example, the brains of patients with Alzheimer disease contain protein aggregates called amyloid plaques that consist primarily of a single polypeptide termed Aβ. This polypeptide is derived from a cellular protein called amyloid precursor protein (APP) through the action of specific proteases. Polypeptide Aβ is prone to form insoluble aggregates. Despite the difficulties posed by the protein’s insolubility, a detailed structural model for Aβ has been derived through the use of NMR techniques that can be applied to solids rather than to materials in solution. As expected, the structure is rich in β strands, which come together to form extended parallel β-sheet structures (Figure 2.62).
How do such aggregates lead to the death of the cells that harbor them? The answer is still controversial. One hypothesis is that the large aggregates themselves are not toxic but, instead, smaller aggregates of the same proteins may be the culprits, perhaps damaging cell membranes.
Protein modification and cleavage confer new capabilities
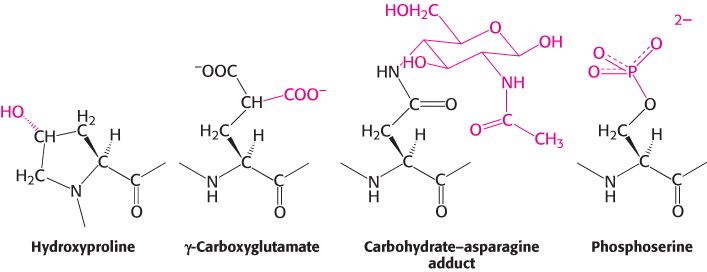
Proteins are able to perform numerous functions that rely solely on the versatility of their 20 amino acids. In addition, many proteins are covalently modified, through the attachment of groups other than amino acids, to augment their functions (Figure 2.64). For example, acetyl groups are attached to the amino termini of many proteins, a modification that makes these proteins more resistant to degradation. As discussed earlier, the addition of hydroxyl groups to many proline residues stabilizes fibers of newly synthesized collagen. The biological significance of this modification is evident in the disease scurvy: a deficiency of vitamin C results in insufficient hydroxylation of collagen, and the abnormal collagen fibers that result are unable to maintain normal tissue strength (Section 27.6). Another specialized amino acid is γ-carboxyglutamate. In vitamin K deficiency, insufficient arboxylation of glutamate in prothrombin, a clotting protein, can lead to hemorrhage (Section 10.4). Many proteins, especially those that are present on the surfaces of cells or are secreted, acquire carbohydrate units on specific asparagine, serine, or threonine residues (Chapter 11). The addition of sugars makes the proteins more hydrophilic and able to participate in interactions with other proteins. Conversely, the addition of a fatty acid to an α-amino group or a cysteine sulfhydryl group produces a more hydrophobic protein.
58
Many hormones, such as epinephrine (adrenaline), alter the activities of enzymes by stimulating the phosphorylation of the hydroxyl amino acids serine and threonine; phosphoserine and phosphothreonine are the most ubiquitous modified amino acids in proteins. Growth factors such as insulin act by triggering the phosphorylation of the hydroxyl group of tyrosine residues to form phosphotyrosine. The phosphoryl groups on these three modified amino acids are readily removed; thus the modified amino acids are able to act as reversible switches in regulating cellular processes. The roles of phosphorylation in signal transduction will be discussed extensively in Chapter 14.
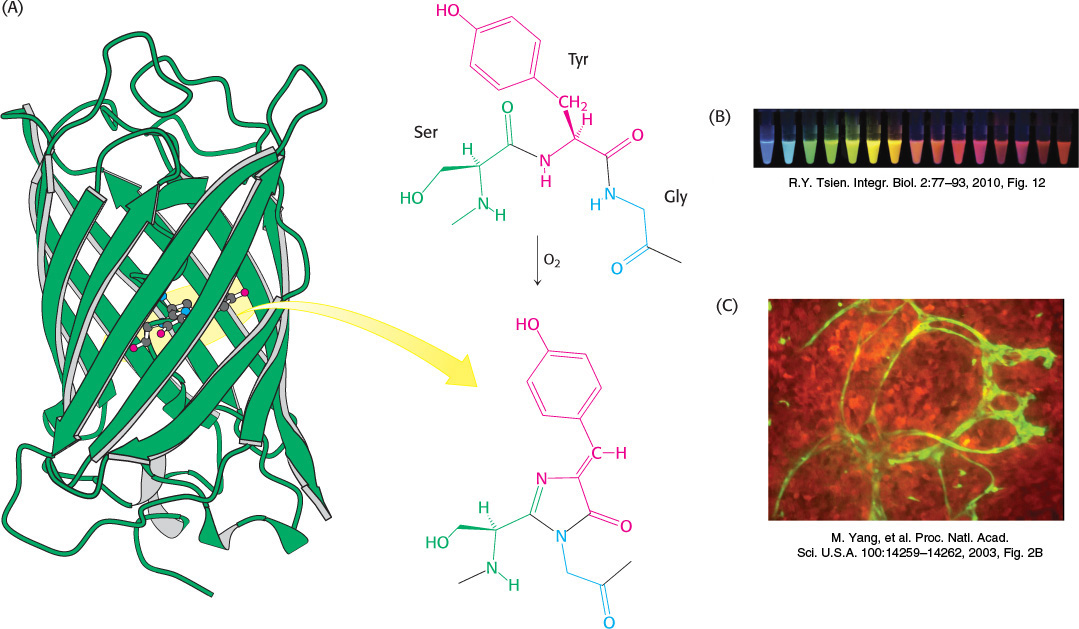
The preceding modifications consist of the addition of special groups to amino acids. Other special groups are generated by chemical rearrangements of side chains and, sometimes, the peptide backbone. For example, the jellyfish Aequorea victoria produces green fluorescent protein (GFP), which emits green light when stimulated with blue light. The source of the fluorescence is a group formed by the spontaneous rearrangement and oxidation of the sequence Ser-
59
Finally, many proteins are cleaved and trimmed after synthesis. For example, digestive enzymes are synthesized as inactive precursors that can be stored safely in the pancreas. After release into the intestine, these precursors become activated by peptide-