36.1The Development of Drugs Presents Huge Challenges
The Development of Drugs Presents Huge Challenges
Many compounds have significant effects when taken into the body, but only a very small fraction of them have the potential to be useful drugs. A foreign compound, not adapted to its role in the cell through long evolution, must have a range of special properties to function effectively without causing serious harm. Let us consider some of the challenges faced by drug developers.
1035
Drug candidates must be potent and selective modulators of their targets
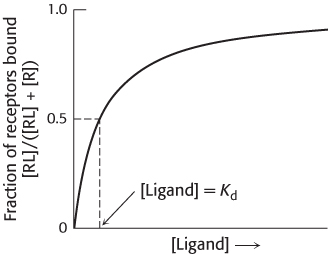
Most drugs bind to specific proteins, usually receptors or enzymes, within the body. To be effective, a drug should bind a sufficient number of its target proteins when taken at a reasonable dose. One factor in determining drug effectiveness is the strength of the interaction between the drug and its target. A molecule that binds to some target protein is often referred to as a ligand. A ligand-
Kd = [R][L]/[RL]
where [R] is the concentration of the free receptor, [L] is the concentration of the free ligand, and [RL] is the concentration of the receptor–
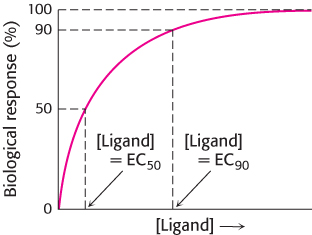
In many cases, biological assays using living cells or tissues (rather than direct enzyme or binding assays) are employed to examine the potency of drug candidates. For example, the fraction of bacteria killed by a drug might indicate the potency of a potential antibiotic. In these cases, values such as EC50 are used. EC50 is the concentration of the drug candidate required to elicit 50% of the maximal biological response (Figure 36.4). Similarly, EC90 is the concentration required to achieve 90% of the maximal response. In the example of an antibiotic, EC90 would be the concentration required to kill 90% of bacteria exposed to the drug. For drug candidates that are inhibitors, the corresponding terms IC50 and IC90 are often used to describe the concentrations of the inhibitor required to reduce a response by 50% or 90%, relative to its value in the absence of inhibitor, respectively.
Values such as the IC50 and EC50 are measures of the potency of a drug candidate in modulating the activity of the desired biological target. To prevent unwanted effects, often called side effects, ideal drug candidates should also be selective. That is, they should not bind biomolecules other than the target to any significant extent. Developing such a drug can be quite challenging, particularly if the drug target is a member of a large family of evolutionarily related proteins. The degree of selectivity can be described in terms of the ratio of the Kd values for the binding of the drug candidate to any other molecules to the Kd value for the binding of the drug candidate to the desired target.
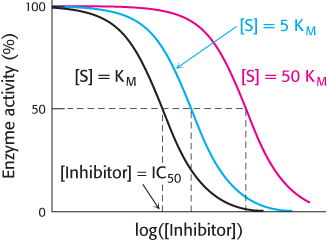
Under physiological conditions, achieving sufficient binding of a target site by a drug can be quite challenging. Most drug targets also bind ligands normally present in tissues; often, the drug and these ligands will compete for binding sites on the target. We encountered this scenario when we considered competitive inhibitors in Chapter 8. Suppose that the drug target is an enzyme and the drug candidate is competitive with respect to the enzyme’s natural substrate. The concentration of the drug candidate necessary to inhibit the enzyme will depend on the physiological concentration of the normal substrate (Figure 36.5). Biochemists Yung-
IC50 = Ki (1 + [S]/KM)
1036
This relation, referred to as the Cheng–
Drugs must have suitable properties to reach their targets
Thus far, we have focused on the ability of molecules to act on specific target molecules. However, an effective drug must also have other characteristics. It must be easily administered and must reach its target at sufficient concentration to be effective. After a drug molecule has entered the body, it is acted upon by several processes—
Administration and absorption. Ideally, a drug can be taken orally as a small tablet. An orally administered active compound must be able to survive the acidic conditions in the gut and then be absorbed through the intestinal epithelium. Thus, the compound must be able to pass through cell membranes at a significant rate. Larger molecules such as proteins cannot be administered orally, because they often cannot survive the acidic conditions in the stomach and, if they do, are not readily absorbed. Even many small molecules are not absorbed well; they may be too polar and not pass through cell membranes easily, for example. The ability to be absorbed is often quantified in terms of the oral bioavailability. This quantity is defined as the ratio of the peak concentration of a compound given orally to the peak concentration of the same dose injected directly into the bloodstream. Bioavailability can vary considerably from species to species, and so results from animal studies may be difficult to apply to human beings. Despite this variability, some useful generalizations have been made. One powerful set consists of Lipinski’s rules.
Lipinski’s rules tell us that poor absorption is likely when
the molecular weight is greater than 500.
the number of hydrogen-
bond donors is greater than 5. 1037
the number of hydrogen-
bond acceptors is greater than 10. the partition coefficient [measured as log(P)] is greater than 5.
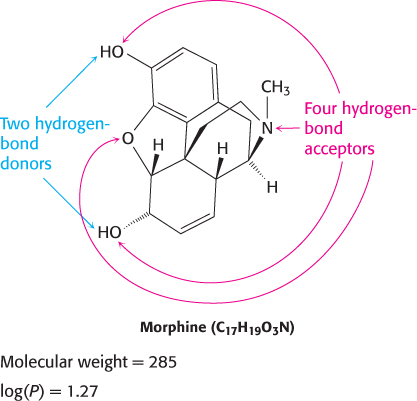
The partition coefficient is a way to measure the tendency of a molecule to dissolve in membranes, which correlates with its ability to dissolve in organic solvents. It is determined by allowing a compound to equilibrate between water and an organic phase, n-octanol. The log(P) value is defined as log10 of the ratio of the concentration of a compound in n-octanol to the concentration of the compound in water. For example, if the concentration of the compound in the n-octanol phase is 100 times that in the aqueous phase, then log(P) is 2. Although the ability of a drug to partition in organic solvents is ideal, because it implies that the compound can penetrate membranes, a log(P) value that is too high suggests that the molecule may be poorly soluble in an aqueous environment.
Morphine, for example, satisfies all of Lipinski’s rules and has moderate bioavailability (Figure 36.7). A drug that violates one or more of these rules may still have satisfactory bioavailability. Nonetheless, these rules serve as guiding principles for evaluating new drug candidates.
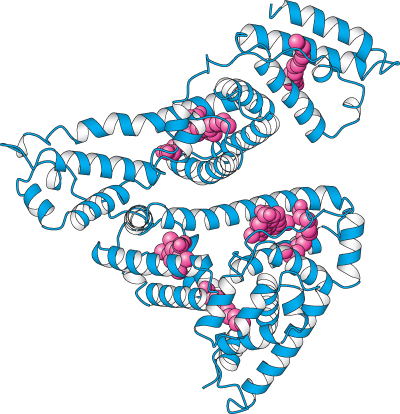
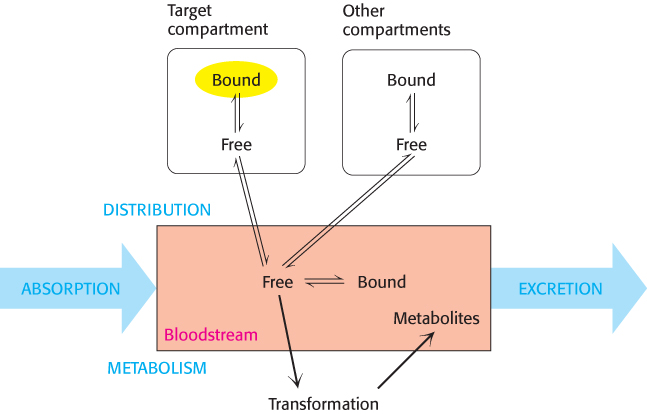
Distribution. Compounds taken up by intestinal epithelial cells can pass into the bloodstream. However, hydrophobic compounds and many others do not freely dissolve in the bloodstream. These compounds bind to proteins, such as albumin (Figure 36.8), that are abundant in the blood serum and by this means are carried throughout the circulatory system.
When a compound has reached the bloodstream, it is distributed to different fluids and tissues, which are often referred to as compartments. Some compounds are highly concentrated in their target compartments, either by binding to the target molecules themselves or by other mechanisms. Other compounds are distributed more widely (Figure 36.9). An effective drug will reach the target compartment in sufficient quantity; the concentration of the compound in the target compartment is reduced whenever the compound is distributed into other compartments.
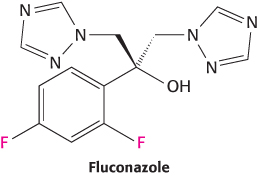
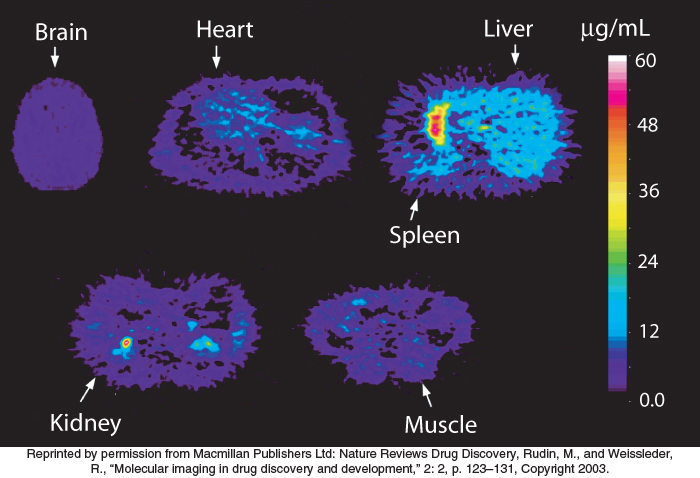
Some target compartments are particularly hard to reach. Many compounds are excluded from the central nervous system by the blood–
Metabolism. A final challenge to a potential drug molecule is to evade the body’s defenses against foreign compounds. Many such compounds (called xenobiotic compounds) are released from the body in the urine or stool, often after having been metabolized to aid in excretion. This drug metabolism poses a considerable threat to drug effectiveness because the concentration of the desired compound decreases as it is metabolized. Thus, a rapidly metabolized compound must be administered more frequently or at higher doses.
Two of the most common pathways in xenobiotic metabolism are oxidation and conjugation. Oxidation reactions can aid excretion in at least two ways: by increasing water solubility, and thus ease of transport, and by introducing functional groups that participate in subsequent metabolic steps. These reactions are often promoted by cytochrome P450 enzymes in the liver (Section 26.4). The human genome encodes more than 50 different P450 isozymes, many of which participate in xenobiotic metabolism. A typical reaction catalyzed by a P450 isozyme is the hydroxylation of ibuprofen (Figure 36.10).
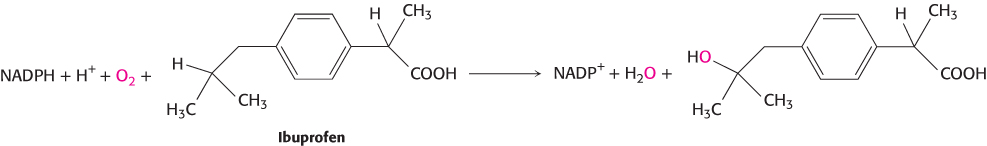
1038
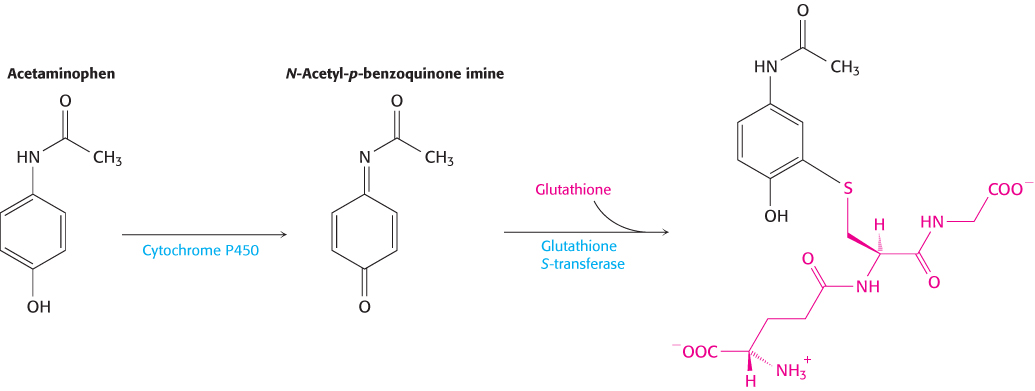
Conjugation is the addition of particular groups to the xenobiotic compound. Common groups added are glutathione (Section 20.5), glucuronic acid, and sulfate (Figure 36.11). These additions often increase water solubility and provide labels that can be recognized to target excretion. Examples of conjugation include the addition of glutathione to the anticancer drug cyclophosphamide, the addition of glucuronidate to the analgesic morphine, and the addition of a sulfate group to the hair-
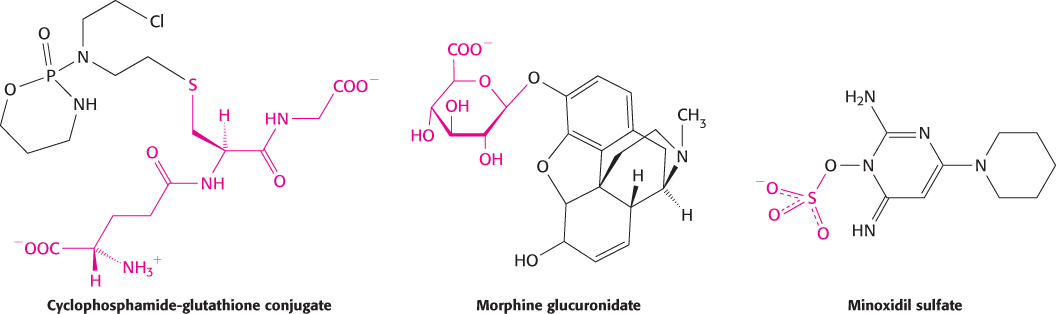
Interestingly, the sulfation of minoxidil produces a compound that is more active in stimulating hair growth than is the unmodified compound. Thus, the metabolic products of a drug, though usually less active than the drug, can sometimes be more active.
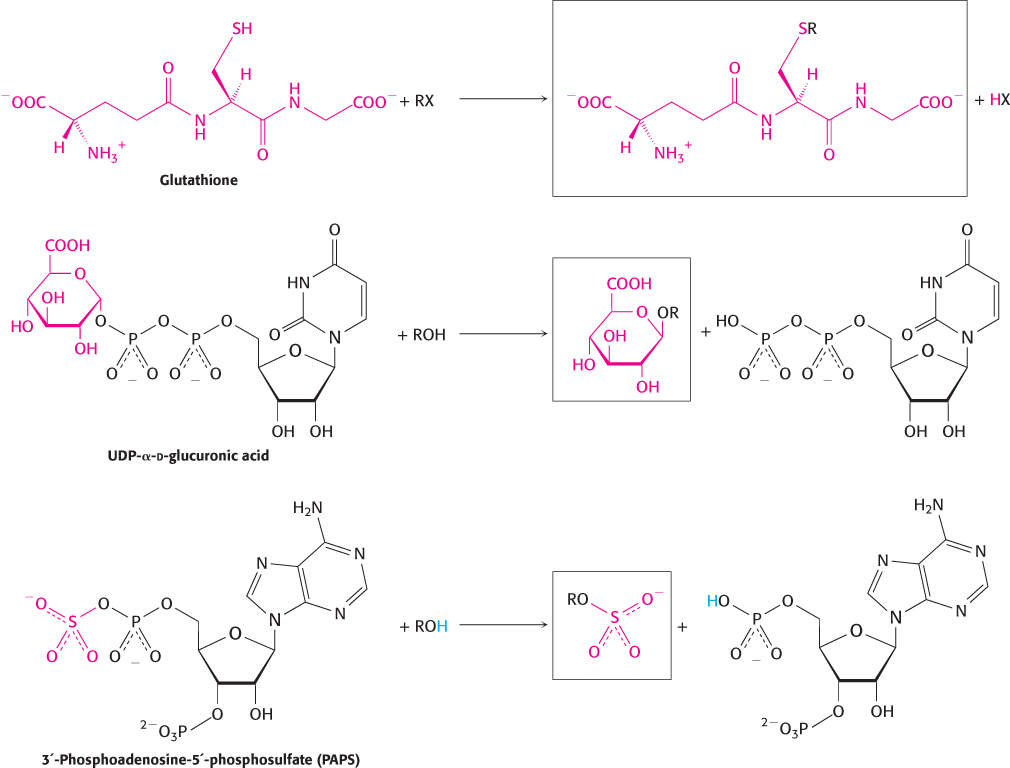
1039
Note that an oxidation reaction often precedes conjugation because the oxidation reaction can generate modifications, such as the hydroxyl group, to which groups such as glucuronic acid can be attached. The oxidation reactions of xenobiotic compounds are often referred to as phase I transformations, and the conjugation reactions are referred to as phase II transformations. These reactions take place primarily in the liver. Because blood flows from the intestine directly to the liver through the portal vein, xenobiotic metabolism often alters drug compounds before they ever reach full circulation. This first-
Excretion. After compounds have entered the bloodstream, they can be removed from circulation and excreted from the body by two primary pathways. First, they can be absorbed through the kidneys and excreted in the urine. In this process, the blood passes through glomeruli, networks of fine capillaries in the kidney that act as filters. Compounds with molecular weights less than approximately 60,000 pass though the glomeruli. Many of the water molecules, glucose molecules, nucleotides, and other low-
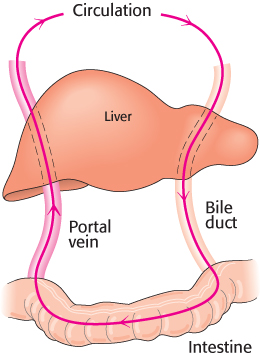
Second, compounds can be actively transported into bile, a process that takes place in the liver. After concentration in the gall bladder, bile flows into the intestine. In the intestine, the drugs and metabolites can be excreted through the stool, reabsorbed into the bloodstream, or further degraded by digestive enzymes. Sometimes, compounds are recycled from the bloodstream into the intestine and back into the bloodstream, a process referred to as enterohepatic cycling (Figure 36.12). This process can significantly decrease the rate of excretion of some compounds because they escape from an excretory pathway and reenter the circulation.
1040
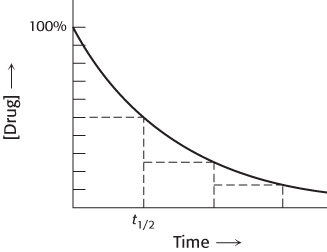
The kinetics of compound excretion is often complex. In some cases, a fixed percentage of the remaining compound is excreted over a given period of time (Figure 36.13). This pattern of excretion results in exponential loss of the compound from the bloodstream that can be characterized by a half-
Toxicity can limit drug effectiveness
An effective drug must not be so toxic that it seriously harms the person who takes it. A drug may be toxic for any of several reasons. First, it may modulate the target molecule itself too effectively. For example, the presence of too much of the anticoagulant drug coumadin can result in dangerous, uncontrolled bleeding and death. Second, the compound may modulate the properties of proteins that are distinct from, but related to, the target molecule itself. Compounds that are directed to one member of a family of enzymes or receptors often bind to other family members. For example, an antiviral drug directed against viral proteases may be toxic if it also inhibits proteases normally present in the body such as those that regulate blood pressure.
A compound may also be toxic if it modulates the activity of a protein unrelated to its intended target. For example, many compounds block ion channels such as the potassium channel hERG (Section 13.4), causing potentially life-
Finally, even if a compound is not itself toxic, its metabolic by-
1041
The toxicity of a drug candidate can be described in terms of the therapeutic index. This measure of toxicity is determined through animal tests, usually with mice or rats. The therapeutic index is defined as the ratio of the dose of a compound that is required to kill one-
Many compounds have favorable properties in vitro, yet fail when administered to a living organism because of difficulties with ADME and toxicity. Expensive and time-