16.2 PRE-mRNA SPLICING AND EDITING
Beyond capping and polyadenylation, eukaryotic transcripts are further processed in a series of reactions referred to as RNA splicing, the splicing of pre-mRNA to produce the mature, translation-competent form of the mRNA. Then, for some eukaryotic mRNAs, the nucleotide sequence is chemically altered by a process known as RNA editing. As we will see, both RNA splicing and RNA editing can expand the coding capacity of the genome by creating mRNAs that are not directly encoded by the DNA. We discuss first the processes of RNA splicing, then RNA editing.
RNA splicing is one of the most important distinctions between gene expression in bacteria and in eukaryotes, and hence has attracted a lot of research interest. Splicing may be a relic of the way that protein-coding sequences originally evolved, providing a glimpse into how modern genes arose. Furthermore, some human diseases, including Duchenne muscular dystrophy and cystic fibrosis, can be caused by aberrant pre-mRNA splicing, suggesting possibilities for therapies if the underlying splicing mechanisms can be understood.
All pre-mRNA splicing mechanisms consist of the ordered breaking and joining of specific phosphodiester bonds to achieve the precise excision of introns. Accurate and efficient splicing relies on base pairing between the pre-mRNA and the splicing machinery to specify the bonds to be broken or formed. In all cases, splicing must be carried out quickly and correctly to produce the mRNAs required for protein production.
A complex of RNA and proteins called the spliceosome is responsible for most pre-mRNA splicing. Compared with ribosomes, spliceosomes are much more dynamic multipiece machines; their assembly occurs at pre-mRNA splice sites and determines whether a given set of splice sites will be used. A small number of introns found in mitochondrial, chloroplast, and bacteriophage transcripts are self-splicing, catalyzing their own excision from a primary transcript without help from proteins. The discovery of self-splicing introns, along with the discovery of the RNA-mediated catalytic activity of the enzyme ribonuclease P (discussed later in this chapter), led to a profound change in our understanding of modern biology—showing that proteins are not the only biological catalysts in cells. The realization that RNA can function both as a carrier of genetic information and as a catalyst led to the idea of an “RNA world” that might have given rise to modern cells and organisms (see Section 16.5).
Eukaryotic mRNAs Are Synthesized as Precursors Containing Introns
In bacteria, a polypeptide chain is generally encoded by a DNA sequence that is colinear with the amino acid sequence, continuing along the DNA template without interruption until the information needed to specify the polypeptide is complete. Although this might appear to be the most efficient way to encode genetic information, primary transcripts isolated from the nuclei of mammalian cells are much larger than required for simply encoding the proteins they produce. Rapid-radiolabeling experiments showed that most of the RNA in these primary transcripts is degraded before it leaves the nucleus. Independent investigations by Phillip Sharp and Richard Roberts in the late 1970s showed that many eukaryotic protein-coding genes are interrupted by noncoding sequences, the introns. Electron microscopy studies of annealed chromosomal DNA and its corresponding mature mRNA revealed regions of complementarity interspersed with looped-out regions in the DNA that did not base-pair with sequences in the mRNA (Figure 16-6). This was the first indication that genes can include both coding regions and noncoding segments that are removed after transcription. As later became apparent, to form mature mRNA, introns must be removed from precursor transcripts and the remaining protein-coding segments, the exons, covalently connected (Figure 16-7).
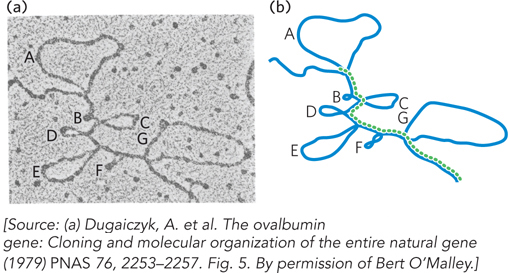
Figure 16-6: A DNA-mRNA hybrid revealing the presence of introns. (a) An electron micrograph and (b) drawing show the ovalbumin gene DNA (blue) hybridized to its mRNA (dashed green line). The DNA contains several loops and regions (A through G) that do not base-pair with the mRNA produced from this region, indicating regions of RNA that are processed out of the mRNA transcript.
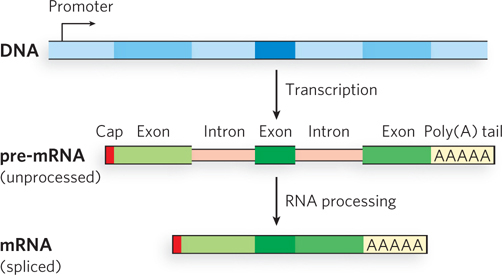
Figure 16-7: Interrupted genes. Introns are non-protein-coding sequences in the DNA and transcribed mRNA that are removed from the RNA during processing to form a contiguous, exon-only, protein-coding mRNA.
Subsequent studies revealed that, in vertebrates, the vast majority of genes contain introns; those that encode histones are among the few exceptions. The occurrence of introns in other eukaryotes varies. Most genes in baker’s yeast, Saccharomyces cerevisiae, lack introns, although they are more common in other yeast species. Introns are also found in a few bacterial and archaeal genes, and even occur within the genes of certain bacteriophages. Although the evolutionary significance of introns is not clear, they can play roles in regulating the amount of mature mRNA produced. For example, some introns include open reading frames that encode regulatory proteins, and others are further processed, after splicing, into small RNAs that base-pair with complementary mRNAs to regulate their stability or translation (see Chapter 22). Splicing also prepares mRNAs to be recognized by proteins that export them from the nucleus and promote their translation by the ribosome.
Introns in DNA are transcribed along with the rest of the gene by RNA polymerases. Then, introns in this primary RNA transcript are excised and the exons are joined to form the mature RNA. A typical mammalian pre-mRNA includes eight introns with an aggregate length 5 to 10 times that of the flanking exons. In general, introns of animal pre-mRNAs vary in size from 50 to 20,000 nucleotides, and most exons are less than 1,000 nucleotides long, with many having just 100 to 200 nucleotides and encoding polypeptide segments of 30 to 60 amino acids. Genes of higher eukaryotes, including humans, have much more DNA devoted to introns than to exons; for example, some human genes have hundreds of introns.
Alternative RNA Splicing Can Generate Multiple Products from a Gene
The transcription of introns might seem to consume cellular resources and energy without returning any benefit to the organism, but introns may confer an advantage. One idea is that introns are vestiges of an ancient molecular parasite not unlike transposons. Another is that introns offer unique advantages to complex organisms because they provide a means of greatly increasing the number of different protein-coding sequences that can be produced, in principle, from a single gene. Comparison of an organism’s genome with the complement of proteins in a given cell type—its proteome—shows that in many cases, the number of different proteins greatly exceeds the number of identified genes. In some cases, at least, this discrepancy is due to alternative splicing, a process in which exons in the primary transcript from a single gene are spliced together in various combinations to produce different mRNAs and thus different polypeptides (Figure 16-8). High-throughput sequencing technology such as RNA-Seq (see Figure 8-12) has revealed that more than 90% of human genes undergo alternative splicing.
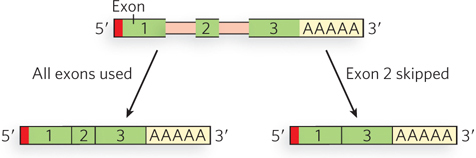
Figure 16-8: Different ways of assembling exons. Alternative splicing can generate multiple products from one gene.

Larry Zipursky
Certain exons are selected for inclusion and others are not, but the order of the exons does not change relative to the primary transcript. In fruit flies, for example, the gene Dscam encodes an immunoglobulin (Ig) superfamily protein, a transmembrane protein required for the formation of neuronal connections, as well as participating in the immune system. Through alternative splicing, Dscam potentially gives rise to 19,008 different extracellular domains linked to one of two alternative transmembrane segments, resulting in 38,016 different possible forms (isoforms) of the Dscam protein! Dscam variants share the same domain structure but contain different amino acid sequences within three Ig domains in the extracellular region. Using Dscam proteins that could be recognized and distinguished by specific antibodies, Larry Zipursky and his colleagues found that each Dscam binds to molecules of the same isoform, but does not bind or binds poorly to other isoforms, contributing to the formation of complex patterns of neuronal connections. Although the Dscam gene is an extreme example, many mammalian genes have two or more alternatively spliced mRNAs derived from the same gene, greatly increasing the complexity of the genome and providing opportunities for regulation at the level of pre-mRNA processing.
Mechanisms of alternative splicing are not yet well understood, but we know that splice sites, nucleotide sequences within the intron and at the borders between introns and exons, play an important role in determining whether an exon is included in the mature mRNA. In some cases, splice sites can be masked in particular cell types, leading to intron skipping and consequent loss of protein production. For example, research on fruit flies in Don Rio’s laboratory showed that in all cells except egg or sperm precursors, the protein Psi binds to a “decoy” splice site adjacent to the true splice site in a gene encoding the enzyme transposase. This prevents proper splicing of the transposase mRNA, thereby preventing transposase production in adult cells where it is not needed and could harm the integrity of the genome by disrupting gene or regulatory sequences.
Regulation by alternative splicing is implicated in some human disease pathways. For example, the inclusion of specific exons in the spliced mRNAs encoding the cell surface molecule CD44 is associated with the progression of certain tumors from localized malignancies to invasive growths Thus, understanding what determines exon choice during alternative splicing might lead to new therapeutic strategies to treat or prevent some cancers.
Complex transcripts can have more than one site for cleavage and polyadenylation, or for alternative splicing patterns, or both. If there are two or more sites for cleavage and polyadenylation, using the one closest to the 5′ end will remove more of the primary transcript sequence. This mechanism, called poly(A) site choice, generates diversity in the variable domains of immunoglobulin heavy chains, which is required for an efficient immune response (see Chapter 14). In fruit flies, alternative splicing patterns produce, from a common primary transcript, three different forms of the myosin heavy chain at different stages of fly development. In rats, both mechanisms (alternative splicing and poly(A) site choice) come into play when a single RNA transcript is processed differently to produce two different hormones: the calcium-regulating hormone calcitonin in thyroid and the calcitonin-gene-related peptide (CGRP) in the brain (Figure 16-9).
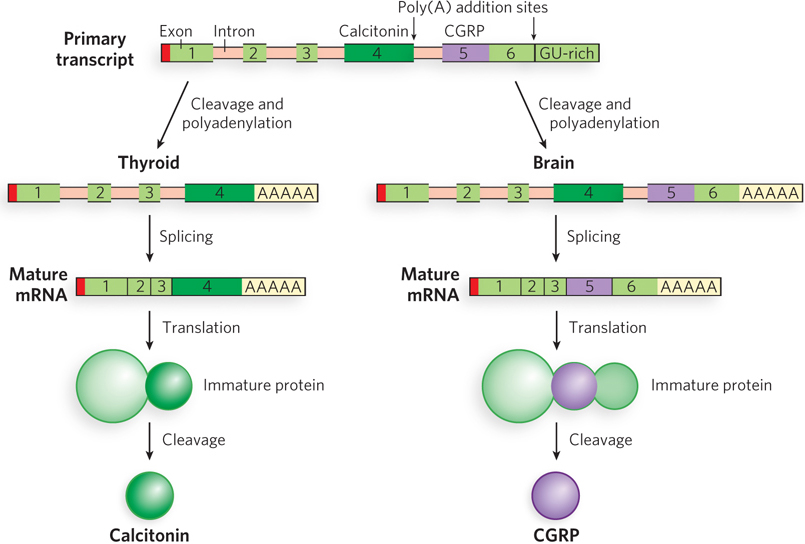
Figure 16-9: Alternative processing of the rat calcitonin gene transcript. Calcitonin and calcitonin-gene-related peptide (CGRP) have completely different protein sequences but are encoded by the same gene. The primary transcript contains two poly(A) addition sites: one used in the thyroid and the other in the brain. In the thyroid, exon 4 is retained; in the brain, exon 4 is eliminated and exons 5 and 6 are retained. The resulting mRNAs encode different polypeptides that are further processed to yield the final hormone products: calcitonin in the thyroid and CGRP in the brain.
The Spliceosome Catalyzes Most Pre-mRNA Splicing
Each intron includes a 5′ splice site, a 3′ splice site, and an internal A residue just upstream of the 3′ splice site, at the branch point (Figure 16-10). Splicing involves a cascade of phosphodiester exchange reactions. First, the 2′-OH of the branch-point A residue attacks the phosphate at the 5′ splice site. Then the 3′-OH of the released exon attacks the phosphate at the 3′ splice site, thus joining the exons and separating them from the intron. These reactions are catalyzed by ribonucleoproteins (RNPs), complexes of non-protein-coding RNAs and proteins. The RNA components of the RNPs base-pair with the mRNA at the 5′ splice site, 3′ splice site, and branch point, positioning the associated proteins for catalysis.
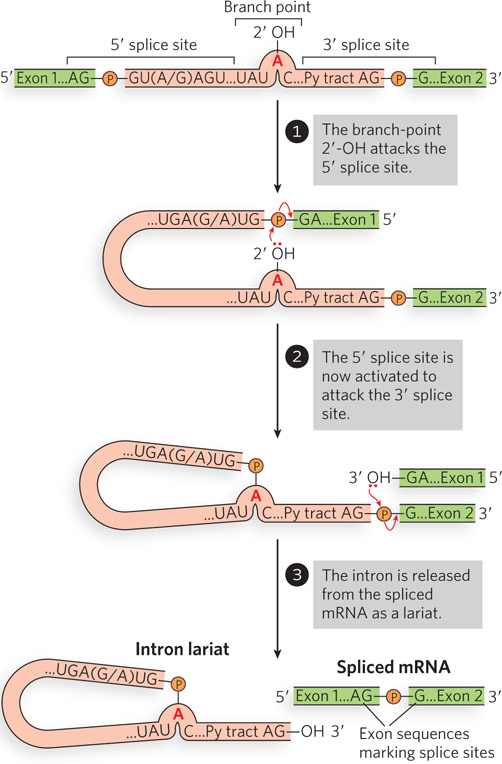
Figure 16-10: An overview of the splicing reaction. Pre-mRNA splicing occurs through two site-specific transesterification reactions that result in phosphodiester bond cleavage and ligation. The 5′ and 3′ splice sites consist of the conserved sequence elements shown; the Py tract in the intron is a string of pyrimidine residues.
Most introns in eukaryotic pre-mRNAs are removed by the spliceosome, a complex of five small nuclear ribonucleoproteins, or snRNPs (pronounced “snurps”), and hundreds of additional protein components. At the heart of each snRNP is a single small nuclear RNA (snRNA) belonging to a class of non-protein-coding eukaryotic RNAs 100 to 200 nucleotides long (Figure 16-11a). Five snRNAs (U1, U2, U4, U5, and U6) involved in splicing reactions are abundant in eukaryotic nuclei. The RNAs and proteins found in snRNPs are highly conserved in eukaryotes from yeast to humans, suggesting that the splicing machinery, and hence introns themselves, were present in the earliest eukaryotes.
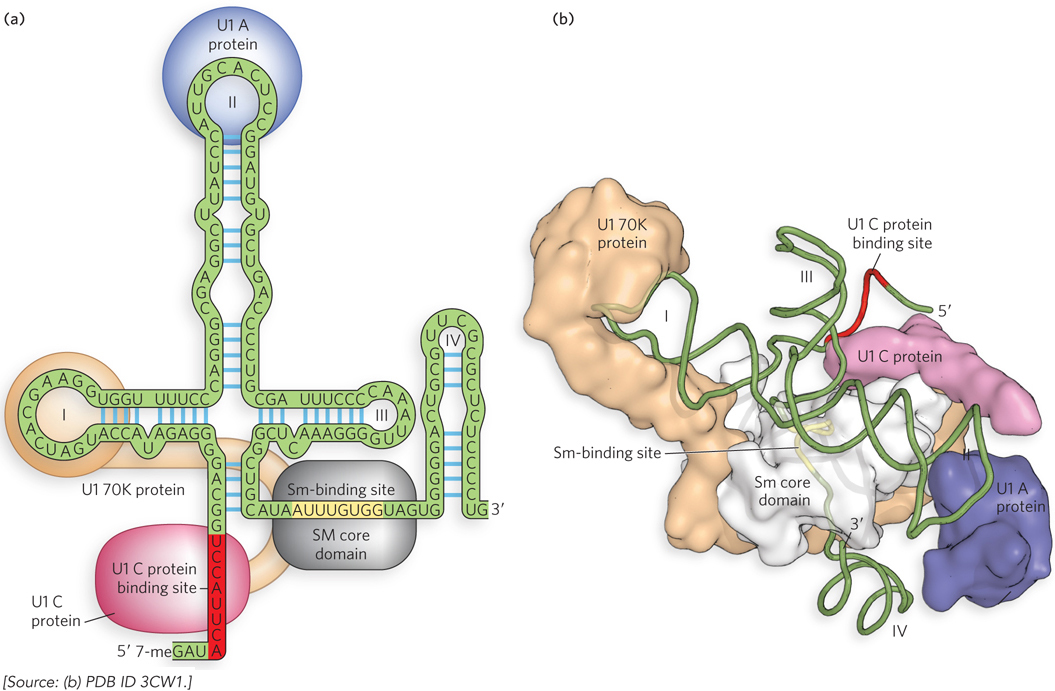
Figure 16-11: The structure of U1 snRNP. Each snRNP consists of an snRNA (U1 is shown here), with an Sm-binding site, as well as unique sequences and secondary structures. In addition, proteins unique to each snRNA bind to form a complex (the U1 snRNP proteins U1 A, U1 C, and U1 70K are shown here). (a) Two-dimensional and (b) three-dimensional representations of the U1 snRNP. Sm proteins bind to the Sm-binding site to form the Sm core domain; unique proteins bind elsewhere in the snRNA to form the complete snRNP.
Each snRNA includes a binding site for a set of proteins—Sm proteins—that are common to all snRNPs (see the How We Know section at the end of this chapter). Seven Sm proteins (SmB, D1, D2, D3, E, F, and G) form a ringlike structure that binds adjacent to a hairpin fold near the 3′ end of the snRNA (see Figure 16-11a). In addition, the snRNA contains sequences that are uniquely recognized by proteins specific to that snRNP. The snRNA–Sm protein complex forms a structure called the Sm core domain. Electron microscopy and x-ray crystallography have been used to visualize individual snRNP proteins and the Sm core (Figure 16-11b), as well as intact snRNPs and assembled spliceosomes (see this chapter’s Moment of Discovery).
Spliceosomes assemble from snRNPs and include other proteins that are not specifically associated with any snRNA. Chief among these are the SR proteins, so named for their serine/arginine-rich region, which play central roles in selecting and regulating splice sites in pre-mRNAs. SR proteins have a common architecture consisting of an RS domain containing repeats of Arg and Ser, and an RNA-recognition motif (RRM). The RS domain is a site of protein-protein interactions that can be enhanced by the phosphorylation of Ser residues. The RRM domain binds to sequences in the pre-mRNA, often within the exons.
One of the key questions to be addressed by biochemical and structural studies is how introns are recognized and cleaved by the spliceosome. Introns have the dinucleotide sequences GU and AG at their 5′ and 3′ ends, respectively, and these sequences mark the sites where splicing occurs. However, these dinucleotide sequences at splice junctions are, by themselves, not sufficiently information-rich to specify splice sites accurately. The surrounding sequences and perhaps the structure of the pre-mRNA itself must play a role in the selection of splice sites, and the ability of snRNPs to bind to these sequences affects the likelihood of splicing. However, the details of how this occurs in the cell are as yet undetermined. Researchers must verify splice sites by comparing the genomic sequence with the corresponding sequence of the mRNA or protein it encodes.
Base pairing between the snRNAs of the spliceosome and the pre-mRNA allows cells to select correct splice sites (Figure 16-12). First, the U1-containing snRNP (referred to simply as U1 snRNP) binds to the GU sequence at the 5′ splice site, along with accessory proteins, including U2AF (U2 auxiliary factor), which binds to the AG and flanking sequences at the 3′ splice site. In an ATP-dependent next step, U2AF recruits U2 snRNP to the branch point. The 2′-OH of the adenosine in the branch point becomes the nucleophile that attacks the phosphodiester bond at the 5′ splice site. Then the U4-U6-U5 trimeric snRNP complex binds, with U6 binding to U2. Next, U1 snRNP is released, U5 snRNP base-pairs with the 5′ exon, and U6 snRNP moves to the 5′ splice site. Once U4 snRNP is released, the U6 and U2 snRNPs catalyze nucleophilic attack of the 2′-OH of the branch-point adenosine on the phosphodiester bond at the 5′ splice site, cleaving the 5′ exon-intron junction and shifting the U5 snRNP to the 3′ splice site.
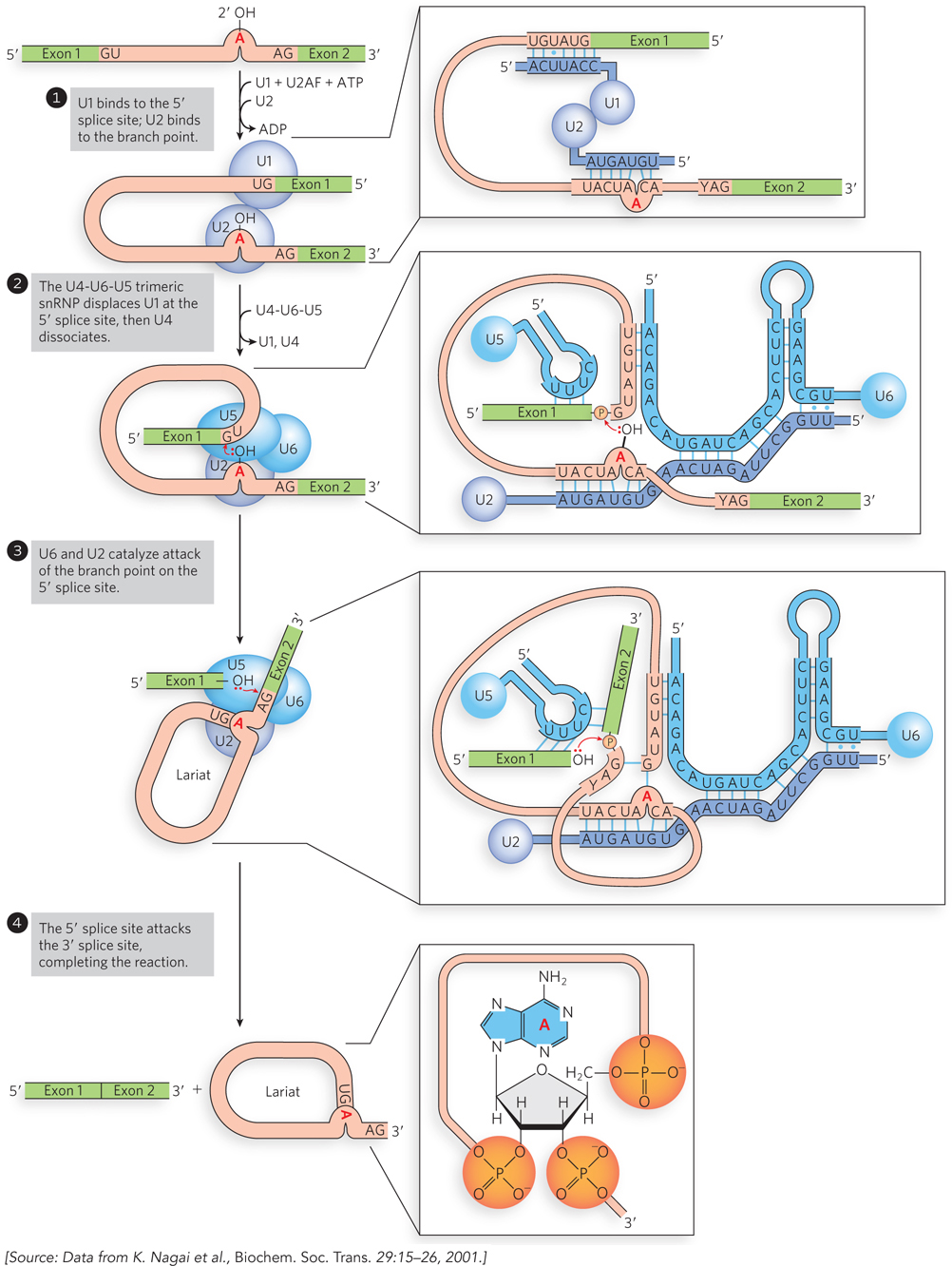
Figure 16-12: Spliceosome assembly on pre-mRNAs involving base pairing to snRNAs. The splicing process is described in the text. In this figure, exons are green; introns, pink; snRNAs, blue. Notice that in step 2, U6 snRNA base-pairs near the 5′ exon binding site where U1 snRNA was formerly bound. As U4 dissociates, the U2-U6-U5 complex remains assembled on the pre-mRNA. U5 base-pairs to both sides of the splice junction to align the RNA for the splicing reaction, and U2 and U6 base-pair to each other.
Formation of the simultaneous 2′ and 3′ phosphodiester linkages at the branch-point adenosine results in a lariat-shaped RNA containing the intron and the 3′ exon. The U2-U6-U5 complex remains bound to the lariat and catalyzes nucleophilic attack of the 3′-OH of the 5′ exon on the phosphodiester bond linking the intron to the 3′ exon, resulting in intron excision with concomitant joining of the 5′ and 3′ exons. ATP hydrolysis is required, presumably for unwinding the spliceosomal RNAs and proteins, as well as helping the snRNAs base-pair with each other and with the pre-mRNA.
Some pre-mRNA introns are spliced by a less common type of spliceosome, in which the U1 and U2 snRNPs are replaced by the U11 and U12 snRNPs. Whereas U1- and U2-containing spliceosomes remove introns with 5′-GU and AG-3′ terminal sequences, the U11-and U12-containing spliceosomes remove a rare class of introns that have 5′-AU and AC-3′ terminal sequences at the intron splice sites. There is no obvious pattern for the use of these so-called AT-AC introns in genes, leading to speculation that they arose in a process parallel to the evolution of the majority of introns.
Some Introns Can Self-Splice without Protein or Spliceosome Assistance

Thomas Cech
Researchers initially assumed that all introns would be removed by protein-catalyzed reactions, but during the early 1980s, Thomas Cech and his coworkers at the University of Colorado discovered that some introns have self-splicing capability. In an in vitro system, Cech and colleagues transcribed an intron-containing piece of DNA isolated from the ciliated protozoan Tetrahymena thermophila, using purified bacterial RNA polymerase. Remarkably, the resulting transcript spliced itself accurately without requiring any Tetrahymena protein enzymes (Figure 16-13). Other RNAs were also found to be capable of functioning as catalysts of RNA processing reactions (see Section 16.5). This exciting discovery was a milestone in our understanding of biological systems. The existence of catalytic RNA molecules implies that RNA competent to both carry and copy genetic information might have formed the chemical basis for early life on Earth, before the evolution of DNA or proteins.
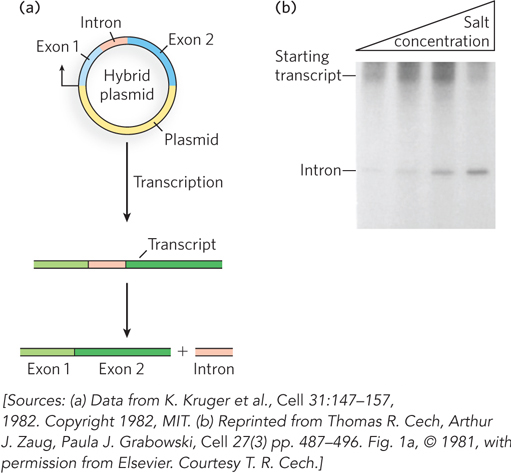
Figure 16-13: Self-splicing of the transcripts of the Tetrahymena large rRNA gene in vitro. (a) A DNA fragment including the intron (red) and flanking sequences (blue) was cloned into a plasmid, which was then transcribed in a test tube by purified RNA polymerase. (b) Samples of pre-rRNA purified from this transcription reaction were incubated in buffer solutions containing sodium and magnesium salts. The pre-rRNA was capable of self-splicing to release the intron, as shown in this agarose gel. The (shorter) intron RNA moves through the gel matrix faster than the (longer) starting transcript. Increasing salt concentration (shown at the top) facilitates the splicing reaction because salts stabilize the folded, catalytically active form of the RNA.
Subsequent research in many laboratories revealed the existence of two distinct classes of self-splicing introns, known as group I and group II. Found mostly in bacteria, organelles, and fungi, group I and group II introns share the ability to self-splice without the involvement of any protein enzymes. Furthermore, neither class requires a high-energy cofactor such as ATP for splicing. It is important to note that group I and group II introns do require proteins for splicing in vivo—not for catalysis, but for forcing the pre-RNA into the correct conformation for splicing to occur.
Group I Introns The catalytic activities and molecular structures of several group I introns, such as the original example discovered in Tetrahymena, have been studied in detail (Figure 16-14). Although the group I splicing reaction requires a guanine nucleoside or nucleotide cofactor, the cofactor is not used as a source of energy. Instead, the 3′-hydroxyl group of the guanosine is the nucleophile in the first step of the splicing pathway (Figure 16-15). The guanosine 3′-OH forms a normal 3′,5′-phosphodiester bond with the 5′ end of the intron. This transesterification reaction releases the 3′ end of the first exon, which then attacks the phosphate of the 3′ splice site, using its 3′-OH in a second transesterification reaction. The intron is released in linear form, with the extra G residue added to its 5′ end. The result is precise excision of the intron and concurrent joining of the exons.
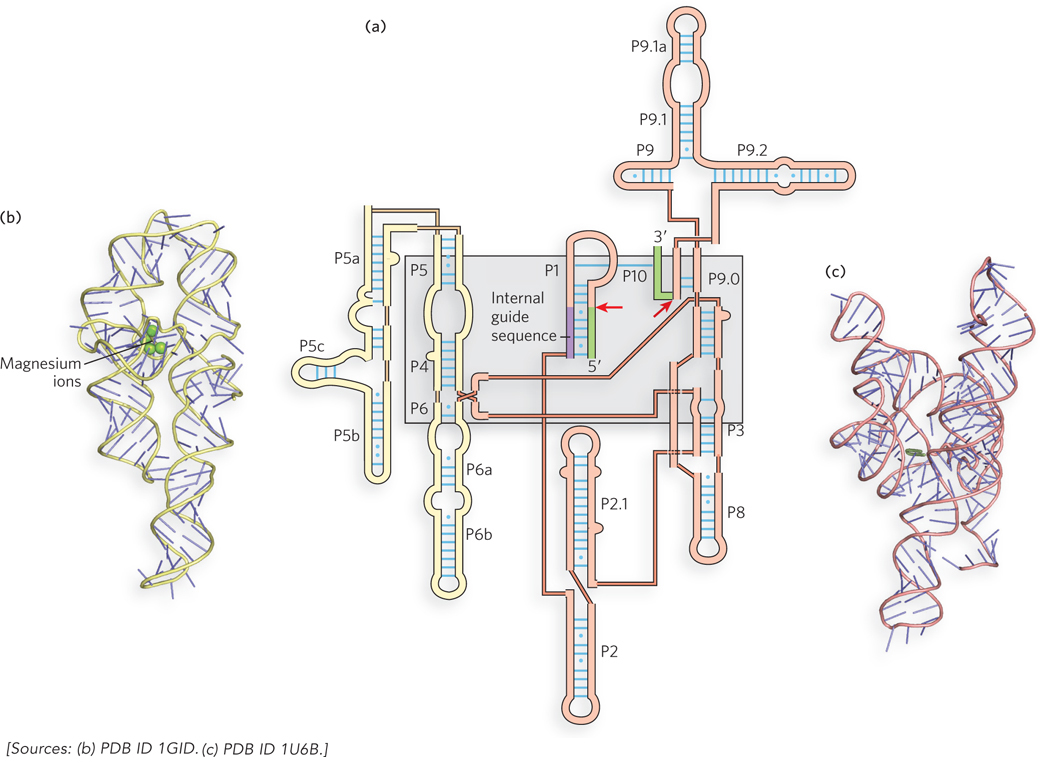
Figure 16-14: Secondary and tertiary structures of group I introns. (a) Group I introns share a common secondary structure in which short segments of the RNA strand fold back on themselves to form base-paired segments (exons shown in green; red arrows indicate splice sites). P segments are base-paired regions, numbered sequentially in the secondary structure; connections between P segments are shown by thinner lines indicating connectivity only, not actual sequence. The gray shaded box indicates parts of the structure that form the catalytic core, or active site, of the intron. (b) The crystal structure of the Tetrahymena group I intron P4-P6 domain (shown in (a) in yellow), the first large RNA structure to be solved, showing how the helices pack together to form a three-dimensional structure. (c) The complete group I intron, which forms an active site for substrate binding and transesterification.
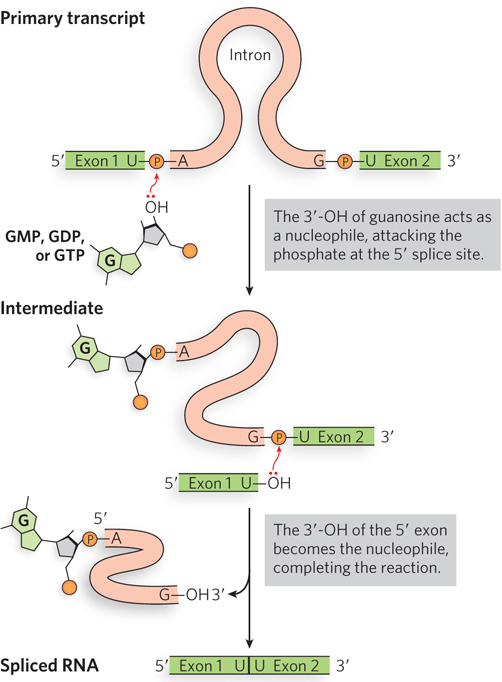
Figure 16-15: The self-splicing mechanism of group I introns. Group I introns use an exogenous guanosine (of a guanine nucleotide) to initiate two transesterification steps in self-splicing. The group I intron is released as a linear structure.
Self-splicing group I introns share several other properties with enzymes, besides accelerating the reaction rate, including their kinetic behavior and specificity. The intron is precise in its excision reaction, largely due to a short sequence—the internal guide sequence—that can base-pair with exon sequences near the 5′ splice site. This pairing promotes the alignment of specific bonds to be cleaved and rejoined.
Because the intron itself is chemically altered during the splicing reaction—its ends are cleaved—it initially appeared to lack one key enzymatic property: the ability to catalyze the same reaction in multiple substrate molecules. In vivo, the intron (414 nucleotides) from Tetrahymena rRNA is quickly degraded after its excision. Experiments have shown, however, that in vitro the intron can act as a true enzyme. A series of intramolecular cyclization and cleavage reactions in the excised intron remove 15 to 19 nucleotides from its 5′ end. The remaining linear RNA promotes nucleotidyl transfer reactions in which some oligonucleotides are lengthened at the expense of others. Although not known to be important in cells, this capability indicates that the RNA can catalyze RNA polymerization. Such activity hints at the possibility that RNA could catalyze its own replication—a key to the “RNA world” hypothesis (see Section 16.5).
Group II Introns Though they have very few conserved sequences, group II introns share a common secondary structure consisting of six base-paired regions referred to as domains I through VI (Figure 16-16a). Domain I contains the binding sites for the 5′ and 3′ splice sites, and domain VI contains an adenosine that functions as the nucleophile to initiate the splicing reaction. Domain V contains sequences critical for the splicing reaction to occur efficiently.
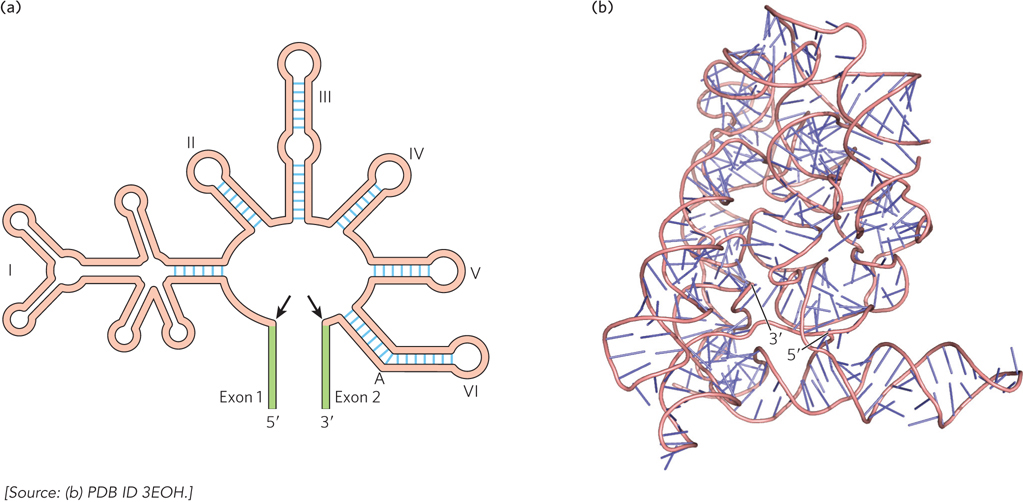
Figure 16-16: Secondary and tertiary structures of group II introns. (a) The overall secondary structure of group II introns is conserved, although many introns contain variable-length insertions in the loops of each base-paired segment. Black arrows indicate the splice sites marking the boundaries of the intron; green segments are the exons. The branch-point A is in domain VI. (b) The crystal structure of a spliced group II intron.
The chemistry of splicing by group II introns is the same as that used by the spliceosome during splicing of eukaryotic pre-mRNAs (Figure 16-17). Here, however, the structure of the RNA itself, rather than the assembly of multiple snRNPs, creates an active site for catalysis, in which the 2′-OH of the branch-point A residue in the intron is directed to attack the phosphodiester bond at the 5′ splice site. As in the spliceosomal reactions, a branched lariat structure forms as an intermediate. After the second step produces the joined exons, the lariat intron can be linearized and degraded or can catalyze further reactions. Thus, like group I introns, group II introns have the ability to catalyze multiple reactions.
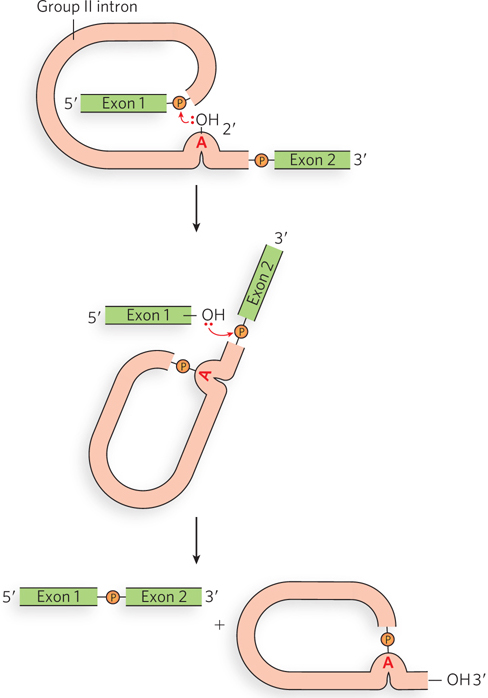
Figure 16-17: Self-splicing mechanism of group II introns. Group II introns self-splice by a pathway similar to that of spliceosome-catalyzed splicing. The group II intron is released as a lariat structure.
The catalytic properties of group II introns have sparked great interest among molecular biologists. The similarity between the mechanisms of self-splicing by group II introns and spliceosome-catalyzed splicing led to the hypothesis that the two processes are evolutionarily related. Perhaps group II introns are renegade spliceosomal RNAs that found a way to survive within genomes as “selfish” genetic elements. Or perhaps group II introns are ancient precursors of the spliceosome, containing just the core RNA components required for catalysis. Either scenario is consistent with the discovery that many group II introns can readily spread to new genes and new organisms by forming a catalytic complex with a protein encoded within the intron itself (Highlight 16-2). Such a mechanism for self-propagation may have enabled a functional subset of spliceosomal RNA to escape from the spliceosome, or it may have maintained the existence of a primitive self-splicing intron long after it could have been supplanted by the spliceosome.
HIGHLIGHT 16-2 EVOLUTION: The Origin of Introns
The origin of introns remains one of the mysteries of modern biology. Because many exons correspond to protein structural domains, it has been argued that introns are relics of early genes, in which sequences were stitched together randomly from shorter segments and only those with useful functions were maintained. According to this line of thinking, bacteria lack introns because competition for rapid growth, and consequent genome streamlining, led to intron loss from all but a few, rare bacterial and bacteriophage genes. An alternative theory is that introns arose more recently on the evolutionary timeline. Analyses of intron positions in related genes from many different species, made possible with the arrival of online whole-genome sequence databases in the 1990s, show that in many cases the introns and their positions within genes are not conserved. This might mean that introns were introduced relatively late in the evolution of modern genomes, or that introns are highly mobile.
The group II introns found in bacteria and in mitochondrial and chloroplast DNA are examples of mobile introns. Like retrotransposons, the introns encode proteins with both endonuclease and reverse transcriptase activities, allowing them to splice themselves back into DNA. In a transposition process termed retrohoming, the encoded protein forms a complex with the intron RNA after the intron is spliced from the primary transcript (Figure 1). Normally, the intron moves from one copy (allele) of a gene to an identical site in another copy of the same gene that lacks the intron. The initial insertion steps reprise the splicing mechanism, but in reverse. Once the RNA strand has been integrated into the DNA, the endonuclease cleaves the opposite DNA strand, and the inserted RNA is copied by the reverse transcriptase function associated with the endonuclease. The RNA is removed and replaced by DNA, converting the RNA intron to an inserted segment of DNA.
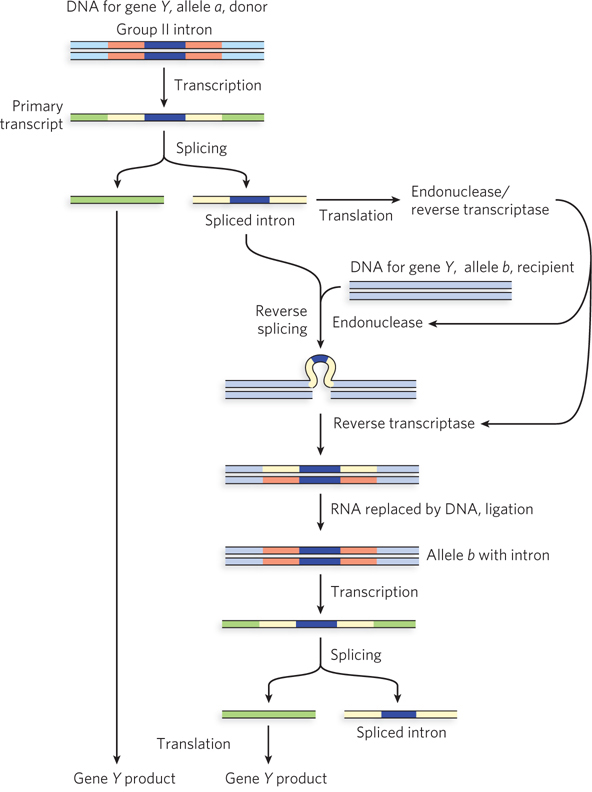
FIGURE 1 Mobile group II introns encode the enzyme activities needed to propagate the intron to new areas of the genome and to new hosts. Two alleles of a gene (Y) differ by the presence (allele a) or absence (allele b) of an intron. In retrohoming, an enzyme called a retrotransposase is produced from a coding sequence within the excised intron. This transposase first cleaves gene Y, allele b DNA at the site of intron insertion, then copies the intron RNA into a cDNA copy that is ligated to create a new, intron-containing allele of gene Y.
Over time, every copy of a particular gene in a population may acquire the intron. Much more rarely, the intron may insert itself into a new location in an unrelated gene. If this event does not kill the host cell, it can lead to the evolution and distribution of an intron in a new location. These mobile group II introns are thought to be the evolutionary precursors of the more widespread (and nonmobile) group II introns found in many eukaryotic genes.
Although spliceosomal introns seem to be limited to eukaryotes, the self-splicing intron classes are not. Genes with group I and II introns have also been found in bacteria and bacterial viruses. Bacteriophage T4, for example, has several protein-coding genes containing group I introns. Group I introns are found in some nuclear, mitochondrial, and chloroplast genes coding for rRNAs, mRNAs, and tRNAs. Group II introns are generally found in the primary transcripts of mitochondrial or chloroplast mRNAs in fungi, algae, and plants. This prevalence is perhaps explained by the observation that both groups of introns are mobile genetic elements, capable of moving between bacterial strains and species (see Chapter 14).
Exons from Different RNA Molecules Can Be Fused by Trans-Splicing
Primary transcripts are sometimes covalently linked to a separate piece of RNA as introns are removed, a process referred to as trans-splicing. Although not known to occur in humans and most other eukaryotes, trans-splicing is the predominant mechanism of mRNA maturation in nematode worms. Genomic sequence data from several different organisms show that trans-splicing is evolutionarily conserved in the nematode family. Maturation of primary transcripts involves trans-splicing of a short leader sequence called SL1 or SL2 onto the 5′ end of the coding sequence for each individual gene (Figure 16-18).
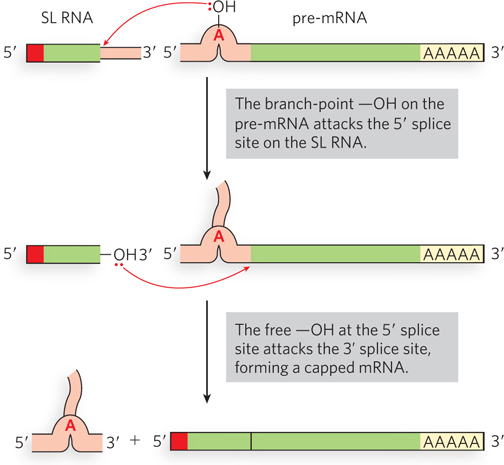
Figure 16-18: Trans-splicing. In nematodes, a short, 5′-capped leader sequence (SL RNA) is spliced onto a primary transcript (pre-mRNA) to produce the mature mRNA.
The mechanism resembles those previously described (designated cis-splicing), using two transesterification steps. The SL (spliced leader) is donated by SL RNA (100 nucleotides). The 5′ splice site is on the SL RNA, and the site of SL addition, the trans-splice site, is the 3′ splice site on the pre-mRNA. The reaction proceeds by way of a branched intermediate similar to the lariat of cis-splicing. Trans-splicing is catalyzed by spliceosomes, including U2, U4, U5, and U6 snRNPs, but not U1. The spliced leader sequence is not translated into protein, but instead plays a regulatory role in coordinating gene expression. In Caenorhabditis elegans, the SL tends to be spliced adjacent to the initiation codon, AUG (often immediately next to it), so the SL is thought to play a role in initiating protein synthesis.
RNA Editing Can Involve the Insertion or Deletion of Bases
Unlike the RNA processing reactions discussed thus far, RNA editing mechanisms seem to have been acquired in recent evolutionary history and to have arisen independently in different groups of organisms. There are two types of RNA editing: insertion or deletion, using guide RNA as a template to add or remove bases from the mRNA, and substitution, in which one base is exchanged for another.
A particularly dramatic example of RNA editing occurs in the mitochondrial pre-mRNAs of trypanosomes, parasitic protozoa that cause human diseases such as sleeping sickness. The pre-mRNAs are edited after synthesis by an enzyme that inserts some U residues and deletes others. Like other kinds of RNA processing, editing of trypanosomal pre-mRNAs was initially discovered by comparing mtDNA sequences with corresponding mRNA sequences. RNA editing activity can be detected by isolating RNA from cells harvested at different times, converting the RNA to DNA with reverse transcriptase (see Chapter 14), then producing many copies of the DNA by the polymerase chain reaction. Comparing the sizes of DNA fragments by gel electrophoresis provides a measure of the relative amounts of edited and pre-edited targets.
RNA editing by insertion and deletion is catalyzed by the editosome, a complex of at least 16 proteins. A key editosome enzyme, RNA-editing terminal uridylyltransferase (TUTase), catalyzes the uridylate addition reaction. The structure of the TUTase ensures that only U residues are added to the mRNA. Because TUTase is essential for the survival of trypanosomes in the host bloodstream, it is a potential target for drug therapy.
The editosome uses small guide RNAs (gRNAs) that are partially complementary to the pre-mRNA regions to be changed (Figure 16-19a). Guide RNAs (35 to 75 nucleotides) are encoded by the trypanosomal mtDNA. Each gRNA contains three functionally important regions: an “anchor” sequence complementary to a target sequence in the pre-edited RNA, a middle segment containing the editing information, and a 3′terminal oligo(U) (5 to 25 nucleotides) extension. The gRNAs base-pair with the target pre-mRNA, and the sequence differences are copied from the gRNA to the pre-mRNA using the gRNA as a template.
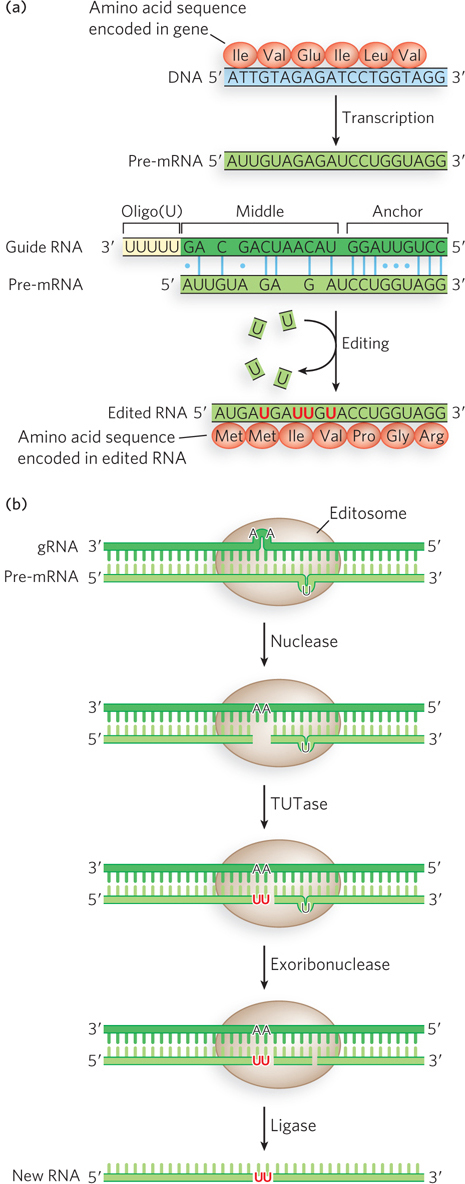
Figure 16-19: RNA editing by nucleotide insertion and deletion. (a) Before editing, a pre-mRNA is missing some U residues required in the mature RNA and contains some extra U residues. A guide RNA (gRNA) in the editosome base-pairs with the pre-mRNA, guiding insertion and deletion of U residues. The insertions and deletions change the protein sequence encoded by the mRNA. (b) The editosome has several enzymatic activities: a nuclease to cleave the pre-mRNA at an insertion site, TUTase to fill in missing U residues, exoribonuclease to delete extra U residues, and ligase to seal the nicks.
The mechanism of the editosome involves an endonucleolytic cut at the mismatch point between the gRNA and the unedited transcript (Figure 16-19b). The insertion step is catalyzed by TUTase, which adds U residues from UTP to the 3′ end of the mRNA. The opened ends are held in place by other proteins in the complex. Another enzyme, a U-specific exoribonuclease, removes any unpaired U residues. After editing has made the mRNA complementary to the gRNA, an RNA ligase rejoins the ends of the edited mRNA transcript. The resulting U insertions or deletions often create a frameshift in the edited mRNA, altering the sequence translated by the ribosome.
RNA Editing by Substitution Involves Deamination of A or C Residues
RNA editing also occurs in human cells and in the viruses that infect them. Many human pre-mRNAs are edited by adenosine deaminase acting on RNA (ADAR), a fascinating enzyme that catalyzes the conversion of adenosine to inosine by removal of the amino group at C-6 of the adenine ring. Typically, ADAR converts just a few of the A residues to I residues within a very large transcript, but those changes are essential for creating the correct sequence to encode a functional protein. Many of the known substrates for ADAR are mRNAs coding for proteins that function in the central nervous system.
An interesting example of ADAR-mediated pre-mRNA editing occurs in the mRNAs encoding glutamate receptor channels (Figure 16-20). These Ca2+ channels, which allow the fast transmission of neural signals, are controlled by L-glutamate, the principal excitatory neurotransmitter in the brain. Two related classes of glutamate receptor channel proteins were found to differ by only a single amino acid, either an Arg or a Gln residue in a defined position of the putative channel-forming segment. However, the genomic DNA sequence encoding the channel segment of both proteins has a glutamine codon (CAG). Researchers discovered that in one set of mRNAs, ADAR converts the A of the CAG codon into an I. Because I can base-pair with C, the A-to-I conversion makes the CIG codon appear to be CGG when detected by standard sequencing reactions involving reverse transcription into DNA. Furthermore, the CIG codon is translated as a CGG codon during protein synthesis. Given that CGG encodes arginine, this editing event results in a glutamine-to-arginine substitution in the glutamate receptor channel protein. Such a change might seem subtle, but in fact the replacement of Gln by Arg in this segment profoundly alters the properties of ion flow across the channel.
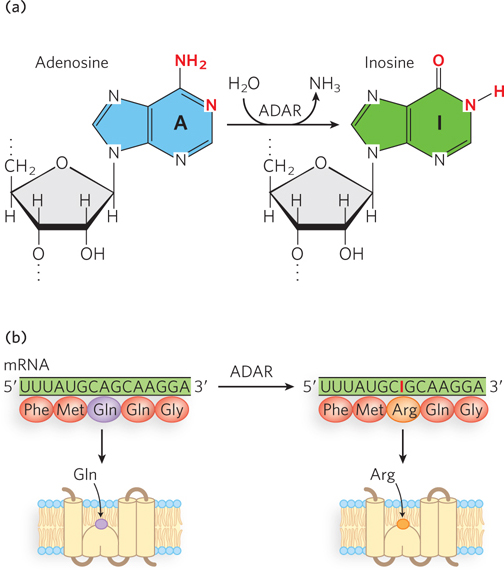
Figure 16-20: A-to-I editing of the mRNA for the glutamate receptor channel protein. (a) ADAR catalyzes the deamination of adenosine to inosine. (b) A-to-I editing results in replacement of a glutamine with an arginine in the protein product. The Arg residue at this position results in a protein that is much more efficient for Ca2+ transport.
A-to-I editing of pre-mRNAs, including the glutamate receptor channel pre-mRNA, often occurs at sites near exon-intron boundaries. This is because ADAR recognizes double-stranded RNA, such as the short duplexes formed in a pre-mRNA where an intron sequence folds back to base-pair with a complementary exon sequence. For this reason, editing necessarily occurs before the pre-mRNA is spliced. The C-terminal domain of Pol II enhances editing by preventing premature splicing, which would otherwise remove the intron recognition sites for ADAR. In some cases, editing occurs within longer regions of duplex RNA, including the large hairpin precursors of regulatory RNAs known as microRNAs (see Section 16.4) and duplexes arising from transcription of double-stranded viral RNAs or base-paired repetitive sequences in the genome. These longer duplex RNAs can be extensively edited, with up to 50% of the adenosines converted to inosines. Sequences or structural properties of double-stranded RNA are recognized by ADAR and help specify the correct editing sites. How such editing affects RNA function is not yet known.
Another class of RNA editing enzymes, the cytidine deaminases, catalyze C-to-U conversions in mRNA substrates. Found in organisms ranging from bacteria to humans, cytidine deaminases can be critical for producing functional mRNAs. Editing involves deamination of a cytosine, converting it to uracil. Such C-to-U editing can be crucial for gene expression. For example, cytidine deamination regulates expression of the apolipoprotein B (ApoB) gene in humans. ApoB is the primary apolipoprotein in low-density lipoprotein (LDL, or “bad” cholesterol), which carries cholesterol in the bloodstream. The full-length form of the protein, ApoB100, is produced in the liver. In the intestine, however, a CAA codon in the mRNA is edited to UAA, creating a stop codon that truncates the protein during translation to a shortened form, ApoB48, which is active in the gut (Figure 16-21). The truncated version lacks a C-terminal domain, affecting the way cholesterol is metabolized in these tissues.
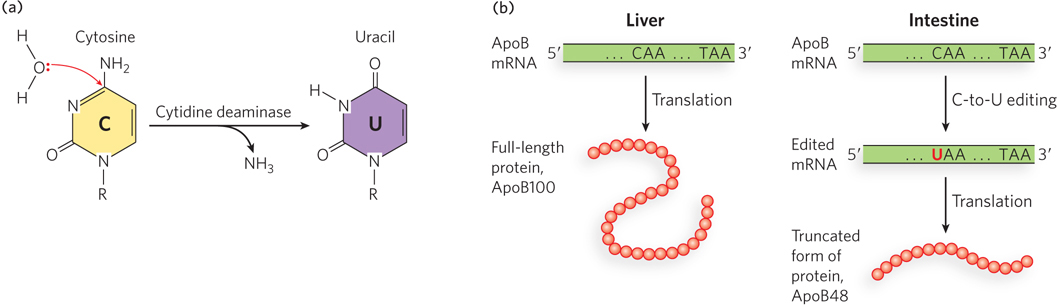
Figure 16-21: C-to-U editing of the mRNA for ApoB protein. (a) Cytidine deaminase catalyzes the deamination of cytidine to uridine. (b) The ApoB gene is expressed in the liver and intestine. In the intestine, the mRNA goes through an additional processing step (after splicing) in which a C residue is changed to a U residue by cytidine deaminase, resulting in a stop codon and producing a smaller protein (ApoB48) that aids in the intestinal absorption of lipids.
Some members of the cytidine deaminase enzyme family are found only in primates. These enzymes, called APOBECs, catalyze C-to-U conversions not only in cellular pre-mRNAs but in viral RNA. Some APOBEC enzymes protect human cells from infection by the human immunodeficiency virus (HIV), by editing and thereby inactivating viral RNA replication intermediates.
SECTION 16.2 SUMMARY
A few bacterial and most eukaryotic pre-mRNAs contain introns, intervening sequences that are removed as the flanking exon sequences are joined together in the process of splicing.
Eukaryotic pre-mRNAs can contain dozens or hundreds of introns. In alternative splicing, the removal of different sets of introns can produce a series of different mRNAs from the same initial transcript.
Most introns are removed by the spliceosome, a complex of five small nuclear ribonucleoproteins (snRNPs); sequences within pre-mRNAs mark the exon-intron boundaries.
Some introns are capable of self-splicing without assistance from any proteins; both kinds of self-splicing introns, group I and group II, also occur in bacterial and mitochondrial RNAs.
In some organisms, trans-splicing produces mRNAs in which exon sequences derive from different primary transcripts.
The nucleotide sequence of eukaryotic mRNAs is sometimes altered by RNA editing enzymes. This can have far-reaching effects, such as altering the protein encoded by the mRNA or influencing the regulation of mRNA translation.
One type of mRNA editing involves the insertion or deletion of U residues. Another type involves enzymatic deamination of A or C residues, converting A to I or C to U.