Importing and Saving Structures
Marvin for JavaScript handles MDL Molfiles V2000 (.mol extension) and ChemAxon Marvin Documents (.mrv extension) as input files. However, other file types (SMILES, ChemAxon Extended SMILES, InChi, Name, CML, MDL Molfile V3000, MDL SDfile, and XYZ) can also be imported if the appropriate webservice is available.
Press the Import button on the General toolbar to open the Import pop-up window. The dialog can be opened by pressing the "Ctrl+O" keyboard shortcut, too. Open the structure file in *.mol or *.mrv format with a text editor (e.g., Windows's Notepad), select all, and copy the file contents to the clipboard then paste it into the input area of the Import window. At the top of the window you can select the appropriate file format from a drop-down list. Finally, pressing the "Import" button loads the structure on the canvas.
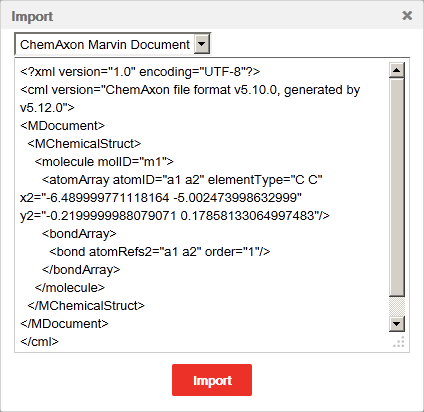
A structure can be saved either in ChemAxon Marvin Document format or in MDL Molfile V2000 format. In order to export in other file formats (SMILES, ChemAxon Extended SMILES, InChi, InChiKey, Name, CML, MDL Molfile V3000, MDL SDfile, and XYZ) the appropriate webservice has to be available.
To save the structures on the canvas, click on the Export button on the General toolbar which opens the Save pop-up window. Alternatively, you can use the "Ctrl+S" keyboard shortcut as well. In this window you can see the canvas contents in the default ChemAxon Marvin Document format. The displayed structure file format can be changed by selecting an option from the drop-down list of supported file formats. To save that to a file you need to copy its content to the clipboard and paste it into a text editor.
Clicking on the lower right corner of the Selection button of the Tools toolbar you can choose between two options:
Rectangle Selection: |
|
Freehand Selection: |
|
Using either selection mode, an atom or a bond can be selected by clicking on it. Please note that in this case the bond gets selected along with the connected atoms. If you want to select a bond without its atoms, you have to choose Freehand Selection, and select the middle of the bond. Alternatively, press the "Ctrl" button and click on the bond, while either selection tool is active.
With Rectangle Selection, you can select an area in rectangle mode while with Freehand Selection the area swept by the red line will be selected.
The background of atoms and bonds will become green when they are selected. In either selection mode, hovering over an atom or a bond, its background becomes green. When you click on this background, the green highlight gets lighter indicating that this part got selected.
Whole molecules can be selected by double-clicking on them. The result is the same as it would be using Rectangle Selection. Whole structures, fragments of molecules, or even several molecules can be selected at the same time.
| • | Separate parts of a molecule or even different molecules can be selected at the same time if the "Shift" button is pressed during selection. |
| • | The "Ctrl+A" keyboard shortcut can also be used to select the whole content of the canvas. |
| • | To clear the selection, you can press the "Esc" button, or simply click on a blank area on the canvas. If there is not any selection on the canvas, pressing the "Esc" button activates the Rectangle Selection tool. |
Once you have made a selection (atom, bond, fragment, or a whole molecule), you can delete it either by clicking on the Delete button on the Tools toolbar or pressing the "Del" button on the keyboard.
Selecting the Delete tool and clicking on an atom or a bond erases said atom or bond. In case of deleting a bond, its atoms will not disappear from the canvas. On the other hand, if you erase an atom, its bonds are also deleted.
The Delete button also works as a rectangle selection tool: click on the button and select the parts you want to erase (the selected parts are highlighted in red). When you release the left mouse button, the highlighted parts will be erased.
Erasing objects is also available from the contextual menu after a right-click on the atom, bond or the selected item you want to delete.
You can clear the whole canvas with one click using the Clear button on the General toolbar or pressing the "Ctrl+Del" keyboard shortcut.
To copy a molecule or a fragment you have to select it first. The Cut and Copy buttons on the General toolbar or in the contextual menu place the content of the selected area on a virtual clipboard. Please note that the copy-paste function is available only within the application. The content of the clipboard can be imported to the canvas with the Paste button on the General toolbar. Their conventional shortcuts, "Ctrl+X", "Ctrl+C", and "Ctrl+V" can be used, too.
Pressing the "Ctrl" button and hovering the cursor over a selected area, you can move a copy of the selected structure by dragging it. When you do this, a "+" sign appears beside the cursor. A copy of the selected structure is placed on the canvas when you release the left button. Note that the new object remains selected when it has been placed on the canvas, so it can be multiplied several times.
You can choose from several options to draw a new atom or to modify an already existing one:
| • | An atom can be inserted by using one of the most frequently used atom buttons on the Atoms toolbar. The chosen atom appears on the tip of the cursor and you can place it on the canvas with a left-click. |
| • | Any of the known elements can be selected from the Periodic Table, the button of which you can find on the Atoms toolbar. Choosing an element, its atomic symbol will appear on the tip of the cursor and you can place it on the canvas several times. At the same time, the Periodic Table window closes automatically. |
| • | Atoms can be placed on the canvas using the keyboard. By typing an atomic symbol, it will appear on the tip of the cursor, and can be placed on the canvas with a left-click or pressing "Enter". Although they are not listed in the Periodic Table, deuterium and tritium symbols (D and T, respectively) can also be used this way. |
| • | When a non-metallic atom is placed on the canvas, it will be automatically completed with implicit hydrogens according to the free valences of the atom. Display of implicit hydrogens depends on the current option in the View Settings dialog box. Metallic elements are exceptions to this rule, they are added to canvas in zero oxidation state. |
| • | Atoms on the canvas can be replaced using either of the above mentioned methods. If you select several atoms at the same time, you can change them simultaneously using either the keyboard or the Periodic System. |
| • | To create a chemical bond between two atoms on the canvas select the appropriate bond type using the Bonds combo button on the Tools toolbar, click on one of the two atoms, and drag the bond to the other atom. |
| • | Selecting a bond type and clicking or dragging the cursor on the canvas will create a new bond with the default length and with carbon atoms on both ends. |
| • | When a bond tool is active and the cursor is over an atom, a grey colored image of a bond in the selected type will appear, ended by a carbon atom. Pressing the "Shift" key will change the bond direction. Click on the atom will place the bond in this position. You can also draw a new bond by dragging from an already existing atom. Just like in the previous case, the length of the bond is the default value, and a carbon atom is on its other end. |
| • | Bond type can be changed by selecting the new bond type from the toolbar and clicking on the bond on the canvas. The bond type can be selected by typing the appropriate shortcut, too. If the cursor is over the canvas, typing a shortcut will select the appropriate bond mode (the Bonds button is activated). However, if the cursor is over an existing bond, typing a shortcut will modify the type of the given bond only. In this case the selected mode (the active button) will not change. |
| • | Note that repeated clicking on a bond will change its type depending on the currently active bond type. If the selected bond type is Single, successive clicks on a bond will change its type according to the Single-Double-Triple-Single-... order. If Double bond type is selected, successive clicks on a bond will alternate its type between Double and Triple. Clicking on a directed bond (Single Up, Single Down, Single Up or Down, or Coordinate bond) will change the bond direction. |
| • | When more than one bonds are selected in a structure, selecting a new bond type will change every selected bond. |
Molecules can be created by connecting atoms with different chemical bonds or with the use of templates from the Templates toolbar.
You can create alkyl or conjugated hydrocarbon chains of arbitrary length using the Chain button on the Tools toolbar. A chain can be created as a new molecule or it can start from an already existing atom on the canvas. After selecting the button you can draw the alkyl chain by dragging or clicking repeatedly on the starting atom. The length of the chain is shown on the tip of the cursor. During drawing, the new chain can be rotated around its starting atom to the desired orientation by dragging. You can create a conjugated polyene chain by pressing the "Ctrl" button when chain-drawing.
Predefined abbreviated groups can also assist you to create complex molecular structures. For selecting an abbreviated group, use the Abbreviated Groups button of the Tools toolbar. When a group abbreviation has been selected from the list of the appearing dialog box, you can decide whether to place it on the canvas in contracted or in expanded form. For the latter option set the Expand checkbox in the dialog. Closing the dialog with the "OK" button will place the group on the tip of the cursor, from where you can put it on the canvas with a left-click (connect it to an already existing structure or start a new molecule). Learn more about abbreviated groups and their handling.
In Marvin for JavaScript you cal also merge separate fragments. Merging is helpful if you want to create ring systems (spiro, fused, or bridged ring systems: one, two, or more than two atoms in common, respectively) or to connect chain-like substructures. For merging two structures, one of them should be selected. When you hover the cursor over the selected fragment, the cursor will change to "move" type (crossed arrows) indicating that the fragment can be moved. Dragging the selected fragment until one or more of its atoms overlap with the "host" molecule atoms will merge the two structures.
Templates can also be merged with a molecule on the canvas. Choose a template from the toolbar (it appears on the tip of the cursor) and move it close to the atoms of the existing structure. When the distance between them has become sufficiently small, the possible merging points will be indicated by blue circles. Left-click will put the template in the merged position.
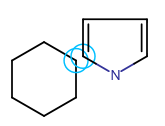
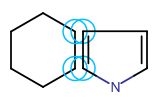
Spiro ring system Fused ring system
Single atoms can be connected to non-terminal atoms in a "sprouted" position. Select an atom symbol, hover the cursor over the atom to which you want to attach the new atom, and press the "Shift" key to see the result of sprouting. Click on the non-terminal atom to attach the new atom to it.
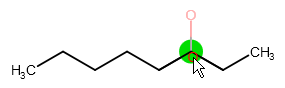
Symmetric templates (cyclohexane, cyclopentane, benzene, pyrrole) can be connected to an atom with a single bond: select the template from the Templates toolbar and hover the cursor over the desired atom to see a grey colored image of the template showing its would-be position. Clicking on the atom will connect the template to the atom with a single bond: one of the implicit hydrogens of the atom will be replaced by the template. Please note that non-symmetric templates (including naphthalene) cannot be sprouted.
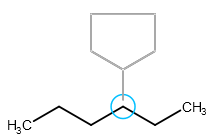
When sprouting, the template and the connecting single bond can be rotated around the connecting atom of the original structure by dragging.
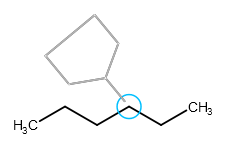
If you want to avoid sprouting, hold down the "Shift" key while clicking on the selected atom. In this case the attaching atom of the template will not replace the connecting atom of the original structure.
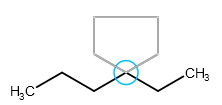
A template can be attached to an already existing bond which is about the same length as the template's bond: clicking on the bond connects the template to the selected bond. The template is rotated automatically in order to fit the chosen bond perfectly.
| o | Pressing the "Shift" button and holding the left mouse key while sprout drawing mirrors the template to the other side of the bond. The side of the bond to place a template can also be altered by dragging the template to the appropriate side. |
| o | When the pyrrole template is selected, and you hover over a bond, pressing the "Space" key will rotate the pyrrole, so you can alter the orientation of the template easily. |
Dragging the selected structure
After selecting them, whole molecules or fragments (parts of molecules) can be moved on the canvas. When you hover the cursor over the selected structure, the cursor "move" cursor will appear indicating that the highlighted part can be dragged on the canvas. If you select a whole molecule, it will be translated without any modifications, but when the selection contains only a fragment, the molecule will be distorted due to the changes in bond lengths and bond angles. Note that the positions of the atoms within the selected fragment are fixed, so only the bonds between the selected and non-selected part of the molecule will be distorted.
When a structure was been selected, it can be rotated around its center in two dimensions.
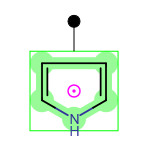
The pink circle with a dot is the center of the selected area (pivot point). The molecule can be rotated around this point by clicking on the green dot and dragging the cursor on the canvas. During dragging, you can see the angle of rotation near this green dot. By default, rotation is done in 6° increments. For continuous rotation hold the "Shift" button while rotating.
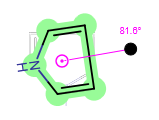
When a whole molecule is selected, the original center of rotation can be relocated by dragging. In this case, if the new rotation center is too close to its original position, it will be snapped back to it. However, pressing the "Shift" key while dragging the center of rotation allows it to be as close to its original place as you wish. Moving the center lets you rotate the molecule around any point of the canvas. The image of the starting structure remains displayed in grey during rotation.
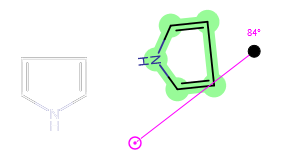
When a selected part is only a fragment which connects to the rest of the molecule with one bond only, you cannot move the pivot point (which is gray in this case). In such cases the rotation center will be that atom of the selected fragment which connects to the non-selected part of the structure.
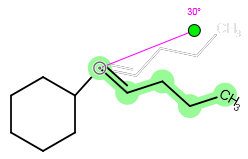
Molecules or fragments can be mirrored either horizontally or vertically using the Mirror Horizontally or Mirror Vertically options in the pop-up menu. For mirroring, you should select the molecule or fragment concerned, right-click inside the selected area, and select the appropriate menu option. Mirroring is possible only for whole, unattached molecules or for fragments which connect with no more than one bond to the unselected part of the structure. In any other case, the mirror options remain inactive.
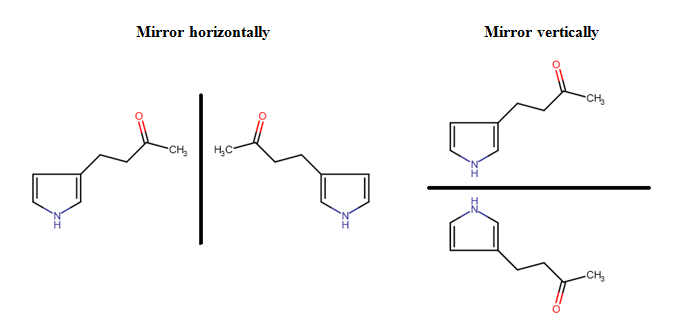
Finally, it has to be mentioned that the rules of undoing/redoing are the same for every kind of transformation: an event begins with pressing down the mouse button and ends with releasing it. The whole action between these two points can be undone or set back in one step.
Currently Marvin for JavaScript supports the display of atomic charges, isotopes, and enhanced stereo specifications as atom properties. All of these attributes can be set in the Atom Properties dialog window available from the pop-up menu after a right-click on the atom.
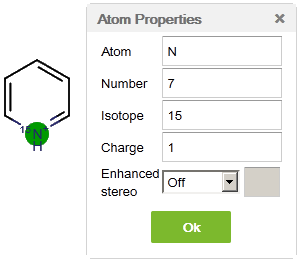
The dialog provides options for setting either new atomic symbols or atomic numbers. Note that these two boxes are synchronized. You can also use this popup window to define atomic properties, such as charge and isotope (mass number). These properties can be set for several atoms at the same time, by selecting the atoms in question and then opening the Atom Properties dialog from the context menu. In this way, the same changes will be applied to every selected atom.
The enhanced stereo notations (Off, Absolute, And, Or) can be added to chiral atoms (marked with wedge bonds) individually in order to assign them to stereogenic groups. The default value is the "Off" identifier, while in case of "Or" and "And" groups a number should accompany the identifier. If you leave the number field empty, number 1 will be associated with the group identifier automatically. Learn more about enhanced stereo specifications in Marvin for JavaScript.
The "OK" button closes the window, and the changes appear in the structure. As a result, a charge will appear as an upper right index of the given atom, while the mass number (isotope type) will be displayed as an upper left index. In case of hydrogen isotopes (deuterium and tritium), their specific atomic symbols can be used (D and T, respectively), or they can be displayed as 2H or 3H, too. If you click on the "[X]" button in the upper right corner of the Atom Properties box or press the "Esc" button, the popup window gets closed without applying any changes to the selected atom.
Note that atomic charges can be modified using the Increase Charge/Decrease Charge buttons on the Tools toolbar, too.
Structure display options can be modified with the View Settings button on the General toolbar, which opens the View Settings dialog box. The options offered here are the following:
| • | Show chiral flag: The visibility of the "Absolute" chiral flag can be turned on/off. |
| • | Index atoms: When checked, the atom indices (the order of the atoms put on the canvas) are displayed. |
| • | Use CPK colors: When checked, the atoms are displayed colored according to the Corey-Pauling-Koltun convention. The two halves of a bond between two atoms will receive the color of the atom they are connected to. |
| • | Show carbon labels: When checked, the carbon atom labels become visible. |
| • | Implicit H: To display the automatically added necessary hydrogens on atoms, an option from the drop-down list in the View Settings window should be chosen. These options are the following: |
Off |
Disable implicit hydrogens on every atom. |
Hetero |
Display implicit hydrogens on hetero atoms. |
Hetero and Terminal |
Display implicit hydrogens on hetero atoms and terminal carbons. (Default setting) |
All |
Display implicit hydrogens on every atom. |