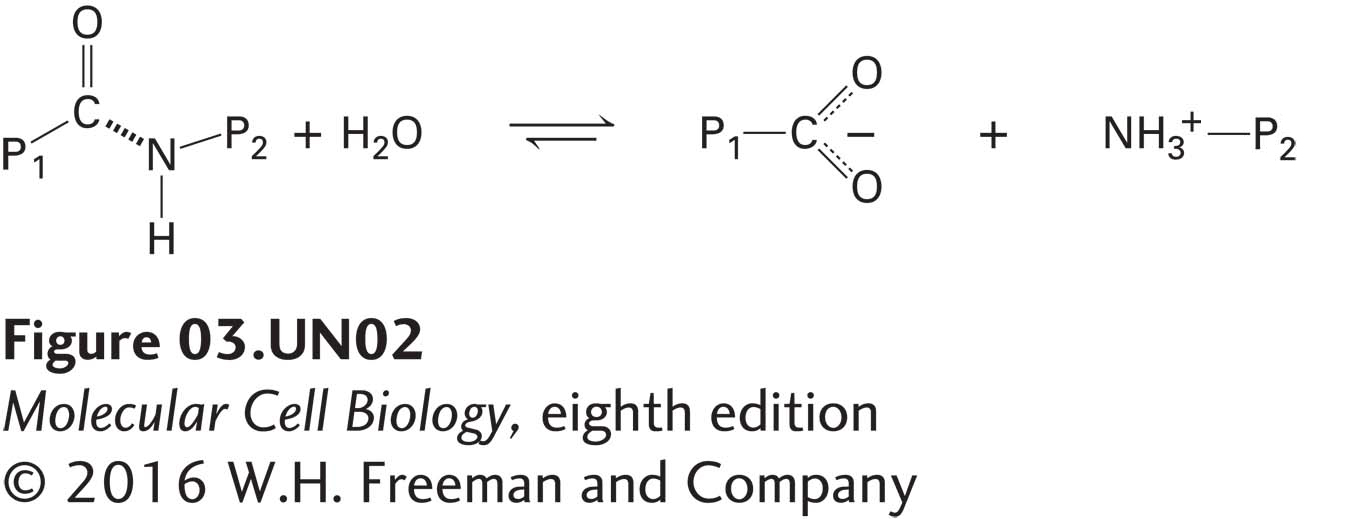
Serine Proteases Demonstrate How an Enzyme’s Active Site Works
Serine proteases, a large family of protein-cleaving, or proteolytic, enzymes, are used throughout the biological world—to digest meals (the pancreatic enzymes trypsin, chymotrypsin, and elastase), to control blood clotting (the enzyme thrombin), even to help silk moths chew their way out of their cocoons (cocoonase). This class of enzymes usefully illustrates how an enzyme’s substrate-binding site and catalytic site cooperate in multistep reactions to convert substrate to product. Here we consider how trypsin and its two evolutionarily closely related pancreatic proteases, chymotrypsin and elastase, catalyze cleavage of a peptide bond in a polypeptide substrate:
Page 93
where P1 is the part of the protein on the N-terminal side of the peptide bond to be cleaved and P2 is the portion on the C-terminal side. We first consider how serine proteases bind specifically to their substrates and then show in detail how catalysis takes place.
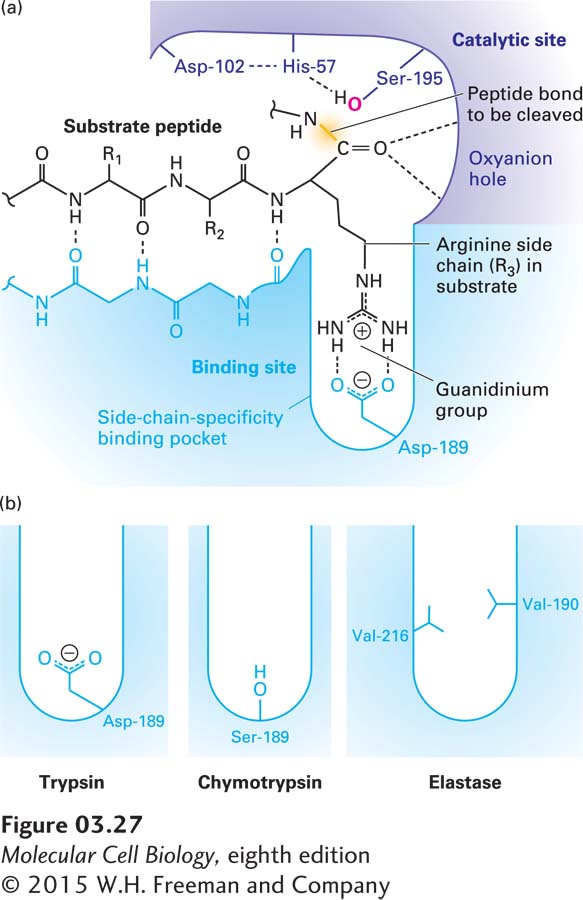
Figure 3-27a shows how a substrate polypeptide binds to the substrate-binding site in the active site of trypsin. There are two key binding interactions. First, the substrate (black polypeptide backbone) and enzyme (blue polypeptide backbone) form hydrogen bonds that resemble those of a β sheet. Second, a key side chain of the substrate that determines which peptide in the substrate is to be cleaved extends into the enzyme’s side-chain-specificity binding pocket, at the bottom of which resides the negatively charged side chain of trypsin’s Asp-189. Trypsin has a marked preference for hydrolyzing substrates at the carboxyl (C ═ O) side of an amino acid with a long, positively charged side chain (arginine or lysine) because the side chain is stabilized in the enzyme’s side-chain-specificity binding pocket by the negative Asp-189.
FIGURE 3-27Substrate binding in the active site of trypsin-like serine proteases. (a) The active site of trypsin (purple and blue molecule) with a bound substrate (black molecule). The substrate forms a two-stranded β sheet with trypsin’s substrate-binding site, and the side chain of an arginine (R3) in the substrate is bound in the side-chain-specificity binding pocket of the binding site. Its positively charged guanidinium group is stabilized by the negative charge on the side chain of the enzyme’s Asp-189. This binding aligns the peptide bond of the arginine appropriately for hydrolysis catalyzed by the enzyme’s active-site catalytic triad (side chains of Ser-195, His-57, and Asp-102). (b) The amino acids lining the side-chain-specificity binding pocket determine its shape and charge and thus its binding properties. Trypsin accommodates the positively charged side chains of arginine and lysine; chymotrypsin, large, hydrophobic side chains such as phenylalanine; and elastase, small side chains such as glycine and alanine. See J. J. Perona and C. S. Craik, 1997, J. Biol. Chem.272:29987–29990.
Slight differences in the structures of otherwise similar binding pockets help explain the differing substrate specificities of two serine proteases related to trypsin: chymotrypsin prefers large aromatic groups (as in Phe, Tyr, Trp), and elastase prefers the small side chains of Gly and Ala (Figure 3-27b). The uncharged Ser-189 in chymotrypsin allows large, uncharged, hydrophobic side chains to bind stably in the binding pocket. The specificity of elastase is influenced by the replacement of glycines in the sides of the binding pocket in trypsin with the branched aliphatic side chains of valines (Val-216 and Val-190), which obstruct the binding pocket (see Figure 3-27b). As a consequence, large side chains in substrates are prevented from fitting into the binding pocket of elastase, whereas substrates with the short alanine or glycine side chains at this position can bind well and be subject to subsequent cleavage.
Page 94
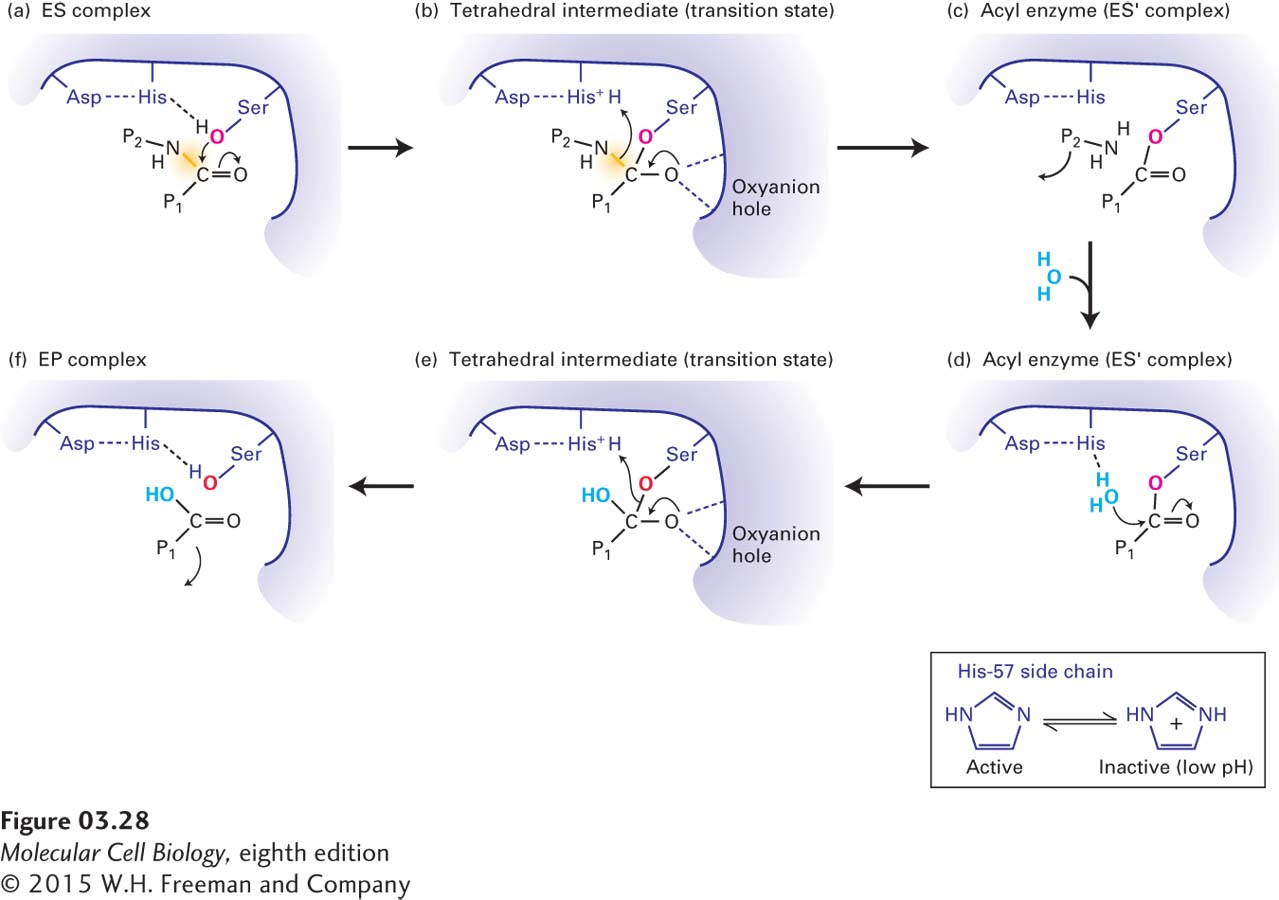
In the catalytic site, all three enzymes use the hydroxyl group on the side chain of a serine at position 195 to catalyze the hydrolysis of peptide bonds in substrate proteins. A catalytic triad formed by the three side chains of Ser-195, His-57, and Asp-102 participates in what is essentially a two-step hydrolysis reaction. Figure 3-28 shows how the catalytic triad cooperates in breaking the peptide bond, with Asp-102 and His-57 supporting the attack of the hydroxyl oxygen of Ser-195 on the carbonyl carbon in the substrate. This attack initially forms an unstable transition state with four groups attached to the carbon (tetrahedral intermediate). Breaking of the C — N peptide bond then releases one part of the substrate protein (NH3 — P2), while the other part remains covalently attached to the enzyme via an ester bond to the serine’s oxygen, forming a relatively stable acyl enzyme intermediate. The subsequent replacement of this oxygen with one from water, in a reaction involving another unstable tetrahedral intermediate, leads to release of the final product (P1 — COOH). The tetrahedral intermediate transition states are partially stabilized by hydrogen bonding with the enzyme’s backbone amino groups in what is called the oxyanion hole. The large family of serine proteases and related enzymes, all of which have an active-site serine, illustrates how an efficient reaction mechanism is used over and over by distinct enzymes to catalyze similar reactions.
FIGURE 3-28Mechanism of serine protease–mediated hydrolysis of peptide bonds. The catalytic triad of Ser-195, His-57, and Asp-102 in the active sites of serine proteases employs a multistep mechanism to hydrolyze peptide bonds in target proteins. (a) After a polypeptide substrate binds to the active site (see Figure 3-27), forming an ES complex, the hydroxyl oxygen of Ser-195 attacks the carbonyl carbon of the substrate’s targeted peptide bond (yellow). Movements of electrons are indicated by arrows. (b) This attack results in the formation of a transition state called the tetrahedral intermediate, in which the negative charge on the substrate’s oxygen is stabilized by hydrogen bonds formed with the enzyme’s oxyanion hole. (c) Additional electron movements result in the breaking of the peptide bond, release of one of the reaction products (NH2 — P2), and formation of the acyl enzyme (ES′ complex). (d) An oxygen from a solvent water molecule then attacks the carbonyl carbon of the acyl enzyme. (e) This attack results in the formation of a second tetrahedral intermediate. (f) Additional electron movements result in the breaking of the Ser-195–substrate bond (formation of the EP complex) and release of the final reaction product (P1 — COOH). The side chain of His-57, which is held in the proper orientation by hydrogen bonding to the side chain of Asp-102, facilitates catalysis by withdrawing and donating protons throughout the reaction (inset). If the pH is too low and the side chain of His-57 is protonated, it cannot participate in catalysis and the enzyme is inactive.
Page 95
The serine protease mechanism points out several key features of enzymatic catalysis. First, enzyme catalytic sites have evolved to stabilize the binding of a transition state, thus lowering the activation energy and accelerating the overall reaction. Second, multiple side chains, together with the polypeptide backbone, carefully organized in three dimensions, work together to chemically transform substrate into product, often by multistep reactions.
[Data from P. Lozano, T. De Diego, and J. L. Iborra, 1997, Eur. J. Biochem. 248:80–85, and W. A. Judice et al., 2004, Eur. J. Biochem. 271:1046–1053.]
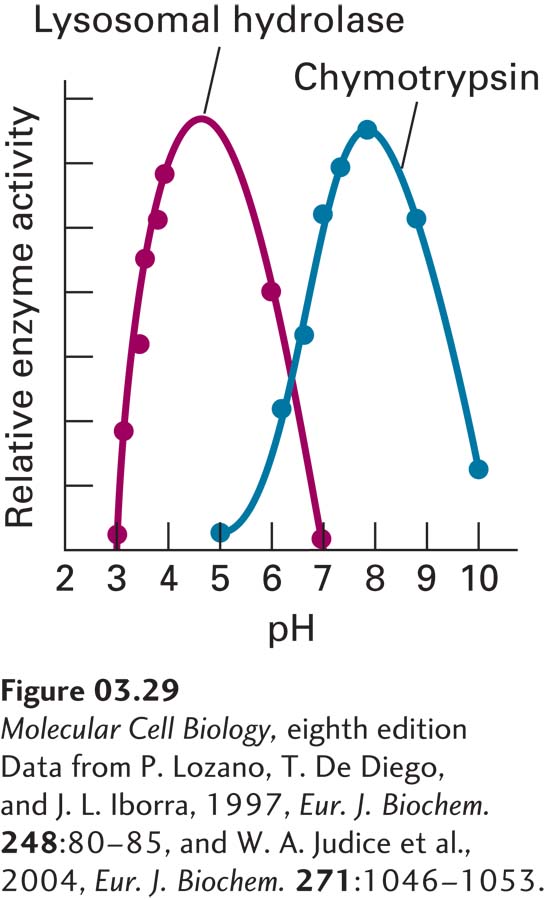
FIGURE 3-29The pH dependence of enzyme activity. In some cases, ionizable (pH-titratable) groups in enzyme active sites or elsewhere in enzymes must be either protonated or deprotonated to permit proper substrate binding or catalysis, or to permit the enzyme to adopt the correct conformation. Measurement of enzyme activity as a function of pH can be used to identify the pKa’s of these groups. The pancreatic serine proteases, such as chymotrypsin, exhibit maximum activity at around pH 8 because of titration of the active-site His-57 (required for catalysis, pKa ~6.8) and of the amino terminus of the protein (required for proper conformation, pKa ~9). Many lysosomal hydrolases have evolved to exhibit a lower pH optimum (~4.5) to match the low internal pH in the lysosomes in which they function.
[Data from P. Lozano, T. De Diego, and J. L. Iborra, 1997, Eur. J. Biochem. 248:80–85, and W. A. Judice et al., 2004, Eur. J. Biochem. 271:1046–1053.]
Third, acid-base catalysis mediated by one or more amino acid side chains is often used by enzymes, as when the imidazole group of His-57 in serine proteases acts as a base to remove the hydrogen from Ser-195’s hydroxyl group. As a consequence, only a particular ionization state (protonated or nonprotonated) of one or more amino acid side chains in the catalytic site may be compatible with catalysis, and thus the enzyme’s activity may be pH dependent. For example, the imidazole of His-57 in serine proteases, whose pKa is ~6.8, can help the Ser-195 hydroxyl attack the substrate only if it is not protonated. Thus the activity of the protease is low at pH <6.8, at which the imidazole is protonated, and the shape of the pH activity profile in the pH range 4–8 matches the titration of the His-57 side chain, which is governed by the Henderson-Hasselbalch equation, with an inflection near pH 6.8 (see chymotrypsin data in Figure 3-29 and see Chapter 2). The activity drops at higher pH values, generating a bell-shaped activity curve, because the proper folding of the protein is disrupted when the amino group at the protein’s amino terminus (pKa ~9) is deprotonated; the conformation near the active site changes as a consequence.
The pH sensitivity of an enzyme’s activity can be due to changes in the ionization of catalytic groups, groups that participate directly in substrate binding, or groups that influence the conformation of the protein. Pancreatic serine proteases evolved to function in the neutral or slightly basic conditions in the intestines; hence their pH optima are ~8. Proteases and other hydrolytic enzymes that function in acidic conditions must employ a different catalytic mechanism. This is the case for enzymes within the stomach (pH ~1), such as the protease pepsin, and for those within lysosomes (pH ~4.5), which play a key role in degrading macromolecules within cells (see the lysosomal hydrolase data in Figure 3-29). Indeed, lysosomal hydrolases, which degrade a wide variety of biomolecules (proteins, lipids, etc.), are relatively inactive at the pH in the cytosol (~7), which helps to protect a cell from self-digestion if these enzymes escape the confines of the membrane-bounded lysosome.
One key feature of enzymatic catalysis not seen in serine proteases but found in many other enzymes is a cofactor or prosthetic group. This “helper” group is a nonpolypeptide small molecule or ion (e.g., iron, zinc, copper, manganese) that is bound in the active site and plays an essential role in the reaction mechanism. Small organic prosthetic groups in enzymes are also called coenzymes. Some of these groups are chemically modified during the reaction and thus need to be replaced or regenerated after each reaction; others are not. Examples include NAD+ (nicotinamide adenine dinucleotide), FAD (flavin adenine dinucleotide) (see Figure 2-33), and the heme groups that bind oxygen in hemoglobin or transfer electrons in some cytochromes (see Figure 12-20). Thus the chemical reactions catalyzed by enzymes are not restricted by the limited number of types of amino acids in polypeptide chains. Many of the vitamins [for example, the B vitamins thiamine (B1), riboflavin (B2), niacin (B3), and pyridoxine (B6), as well as vitamin C], which cannot be synthesized in mammalian cells, function as, or are used to generate, coenzymes. That is why supplements of vitamins must be added to the liquid medium in which mammalian cells are grown in the laboratory (see Chapter 4).
Small molecules that can bind to active sites and disrupt catalytic reactions are called enzyme inhibitors. Such inhibitors are useful tools for studying the roles of enzymes in cells and organisms. Inhibitors that bind directly to an enzyme’s binding site and thus compete directly with the normal substrate are called competitive inhibitors. Noncompetitive inhibitors are those that interfere with enzyme activity in other ways—for example, by binding to some other site on the enzyme and changing its conformation. Enzyme inhibitors complement the use of genetic mutations and a technique called RNA interference (RNAi) for probing an enzyme’s function in cells (see Chapter 6). In all three approaches, the cellular consequences of disrupting an enzyme’s activity can be used to deduce the normal function of the enzyme. The same approaches can be used to study the functions of nonenzymatic macromolecules. Interpreting the results of inhibitor studies can be complicated, however, if, as is often the case, the inhibitors block the activity of more than one protein.
Page 96
Small-molecule inhibition of protein activity is the basis for many drugs as well as for chemical warfare agents. Aspirin inhibits enzymes called cyclooxygenases, whose products can cause pain. Sarin and other nerve gases react with the active serine hydroxyl groups of both serine proteases and a related enzyme, acetylcholine esterase, which is a key enzyme in regulating nerve conduction (see Chapter 22).