CONCEPT10.1 Genetics Shows That Genes Code for Proteins
Following Mendel’s definition of the gene as a physically distinct entity (see Concept 8.1), biologists identified the genetic material as DNA (see Concept 9.1). In this chapter we will show that in most cases, genes code for proteins, and it is proteins that determine phenotypes. The connection between protein and phenotype was made before it was known that DNA is the genetic material.
Observations in humans led to the proposal that genes determine enzymes
The identification of a gene product as a protein began with a mutation. In the early twentieth century, the English physician Archibald Garrod saw several children with a rare disease. One symptom was that the urine turned dark brown or black in air, and for this reason the disease was named alkaptonuria (“black urine”).
Garrod noticed that the disease was most common in children whose parents were first cousins. Mendelian genetics had just been “rediscovered,” and Garrod realized that because first cousins can inherit some alleles that are the same from their shared grandparents, their children are more likely than others to inherit a rare mutant allele from both parents—and therefore are more likely to be homozygous recessive for rare genetic conditions (see Figure 8.8B). Garrod proposed that alkaptonuria was a phenotype caused by a recessive mutant allele.
LINK
You can review the inheritance patterns of recessive alleles in Concept 8.1; see especially Figure 8.8
Garrod took the analysis a step further by identifying the biochemical abnormality in the affected children. He isolated from them an unusual substance, homogentisic acid, which accumulated in the blood, joints (where it crystallized and caused severe pain), and urine (where it turned black when exposed to air).
Enzymes as biological catalysts had just been discovered, and Garrod proposed that in healthy individuals, homogentisic acid might be broken down to a harmless product by an enzyme:
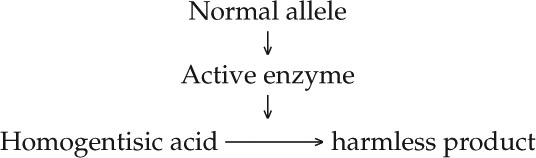
Garrod speculated that the synthesis of the active enzyme is determined by the dominant wild-type allele of the gene that was mutated in alkaptonuria patients. These and other studies led him to correlate one gene to one enzyme, and to coin the term “inborn error of metabolism” to describe this kind of genetically determined biochemical disease.
But Garrod’s hypothesis needed direct confirmation by the identification of the specific enzyme and the specific gene mutation involved. In 1958 the enzyme was identified as homogentisic acid oxidase, which breaks down homogentisic acid to a harmless product, just as Garrod predicted (FIGURE 10.1). The specific DNA mutation leading to alkaptonuria was described in 1996.
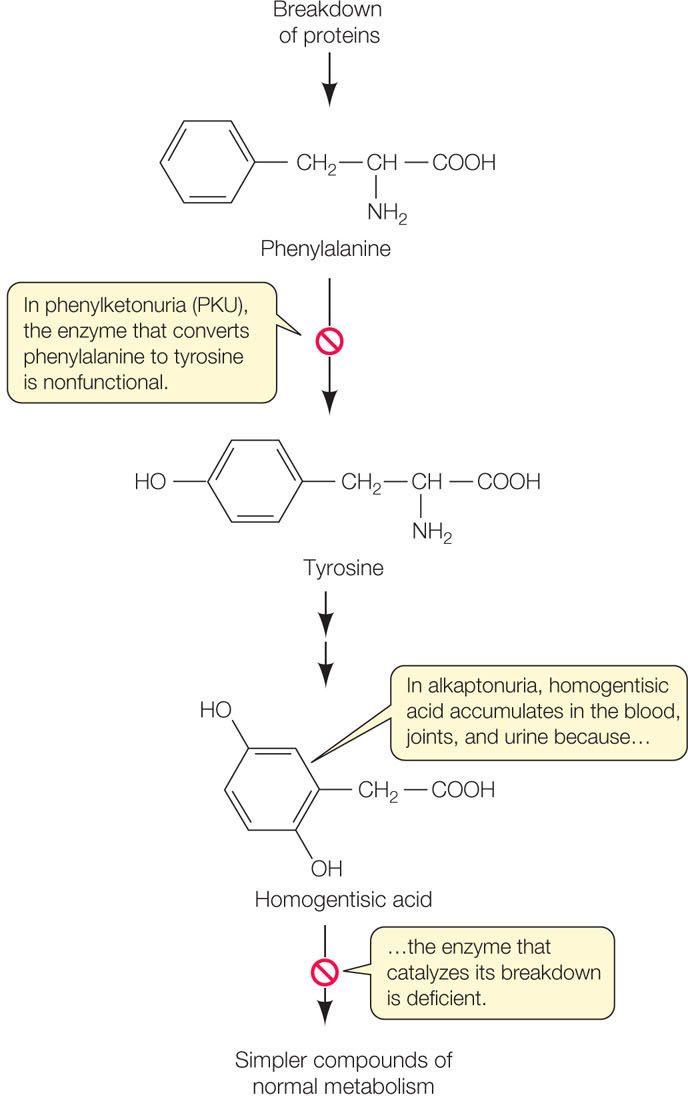
Homogentisic acid is part of a biochemical pathway that catabolizes proteins, with the amino acids phenylalanine and tyrosine as intermediate products. Phenylketonuria, another genetic disease involving the same pathway, was discovered several decades after Garrod did his work. In phenylketonuria, the enzyme that converts phenylalanine to tyrosine is nonfunctional (see Figure 10.1). If left untreated, this disease leads to significant intellectual disability. Fortunately, the accumulation of phenylalanine can be easily detected in the blood of a newborn infant, and if the child consumes a diet low in proteins containing phenylalanine, intellectual disability is avoided.
The concept of the gene has changed over time
The phenotypic expression of mutations underlying alkaptonuria and phenylketonuria led to the “one gene–one enzyme” hypothesis. Once it was known that proteins (including enzymes) are polymers of amino acids, and that the sequence of amino acids determines protein function, it became clear that a mutant phenotype arises from a change in the protein’s amino acid sequence. However, scientists soon realized that the one gene–one enzyme (or one protein) hypothesis was an oversimplification. Once again, studies of human mutations were a key to this realization.
196
Genetics shows that genes code for proteins
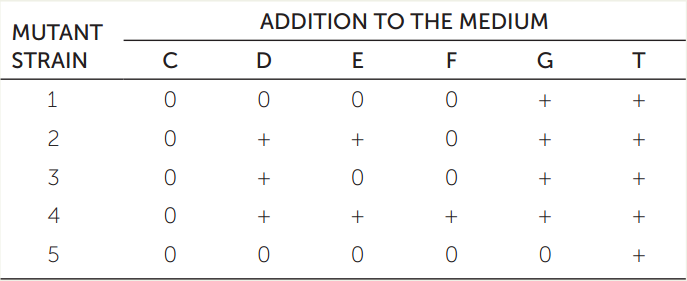
Wild-type bacteria can synthesize the amino acid tryptophan (T) using a biochemical pathway that begins with chorismate (C) and involves four intermediates that we will call D, E, F, and G. Bacterial strains with mutant alleles for the five enzymes (1–5) involved in this pathway cannot synthesize tryptophan, and it must be supplied as a nutrient in the growth medium. The table gives the phenotypes of five mutant strains, each of which has a mutation in a gene for a different enzyme of the five. A “+” means the strain grew when the indicated compound was added to the medium, and a “0” means the strain did not grow. Based on these data, order the compounds (C, D, E, F, G, and T) and the enzymes (1, 2, 3, 4, and 5) in a biochemical pathway, as in the (incorrect) depiction here:

Hint: Mutant strain 5 will not grow if any compound other than T is supplied, so it must carry a loss-of-function mutation in the enzyme that transforms another compound (either C, D, E, F, or G) into T. Thus enzyme 5 is the final enzyme in the pathway.
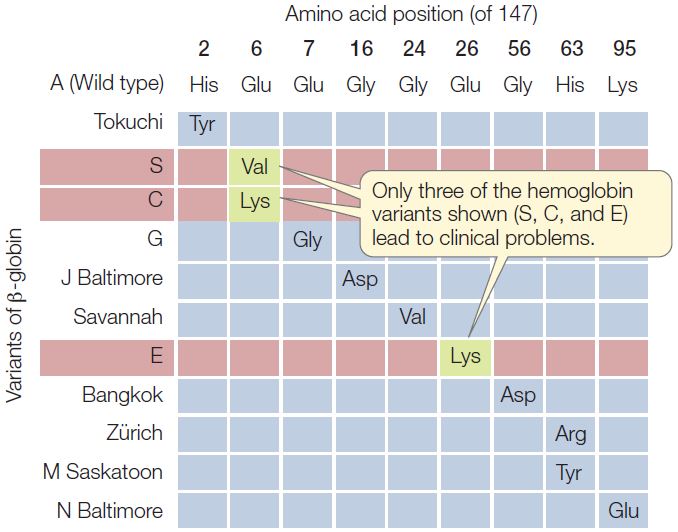
In humans, the oxygen-carrying protein hemoglobin has a quaternary structure of four polypeptide chains—two α-chains and two β-chains (see Concept 3.2). Concept 9.3 introduced sickle-cell disease, which is caused by a point mutation in the gene for β-globin and is inherited as an autosomal recessive (carried on an autosome rather than a sex chromosome). In sickle-cell disease, one of the 147 amino acids in the β-globin chain is abnormal: at position 6, the normal glutamic acid has been replaced by valine. This replacement changes the charge of the protein (glutamic acid is negatively charged and valine is neutral), causing it to form long, needlelike aggregates in the red blood cells. The phenotypic result is anemia, an impaired ability of the blood to carry oxygen.
Because hemoglobin is easy to isolate and study, its variations in the human population have been extensively documented (FIGURE 10.2). Hundreds of single amino acid alterations in β-globin have been reported. For example, at the same position that is mutated in sickle-cell disease (resulting in hemoglobin S), the normal glutamic acid may be replaced by lysine, causing hemoglobin C disease. In this case, the resulting anemia is usually not severe. Many alterations of hemoglobin do not affect the protein’s function. That is fortunate, because about 5 percent of all humans are carriers for one of these variants.
Studies of proteins that are made up of multiple polypeptides (such as hemoglobin) resulted in a modification of the one gene-one enzyme hypothesis. Scientists began to think of the relationship as one gene-one polypeptide, which remains a powerful and useful concept today. However, as you will see in this and later chapters, we are learning that this, too, is an oversimplification.
LINK
Exceptions to the one gene-one polypeptide relationship include the alternative splicing of RNA, which can produce multiple functional polypeptides from a single gene; see Concept 11.4
Mutations such as those that cause alkaptonuria and phenylketonuria result in alterations in amino acid sequences. But not all genes code for polypeptides. As we will see below and in Chapter 11, some DNA sequences are transcribed into RNA molecules that are not translated into polypeptides, but instead have other functions. Like all other DNA sequences, these RNA genes are subject to mutations, which may or may not affect the functions of the RNAs they produce.
197
Our understanding of genes and how they are expressed has increased dramatically over the past 60 years, since Watson and Crick first worked out the structure of DNA (see Concept 9.1). Let’s begin our discussion of gene expression with an overview of the kinds of RNA molecules that are involved in transcription and translation.
Genes are expressed via transcription and translation
Molecular biology is the study of nucleic acids and proteins, and it often focuses on gene expression. As we described briefly in Chapter 3, genes are expressed as RNAs, many of which are translated into proteins. This process involves two steps:
- During transcription, the information in a DNA sequence (a gene) is copied into a complementary RNA sequence.
- During translation, this RNA sequence is used to create the amino acid sequence of a polypeptide.
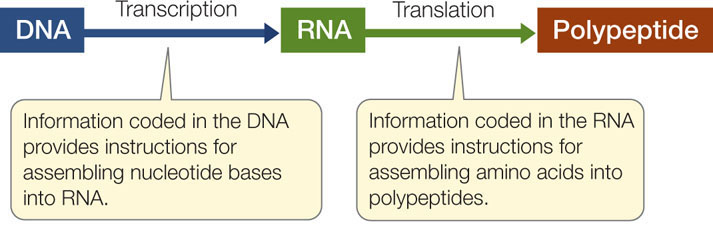
Here we will consider three types of RNA with regard to their roles in protein synthesis:
- Messenger RNA and transcription: When a particular gene is expressed, the two strands of DNA unwind and separate into a coding strand and a template strand. The template strand is then transcribed to produce an RNA strand by complementary base pairing. The RNA strand is then modified to produce a messenger RNA (mRNA). In eukaryotic cells, the mRNA is processed in the nucleus and then moves to the cytoplasm, where it is translated into a polypeptide (FIGURE 10.3). The nucleotide sequence of the mRNA determines the ordered sequence of amino acids in the polypeptide chain, which is built by a ribosome.
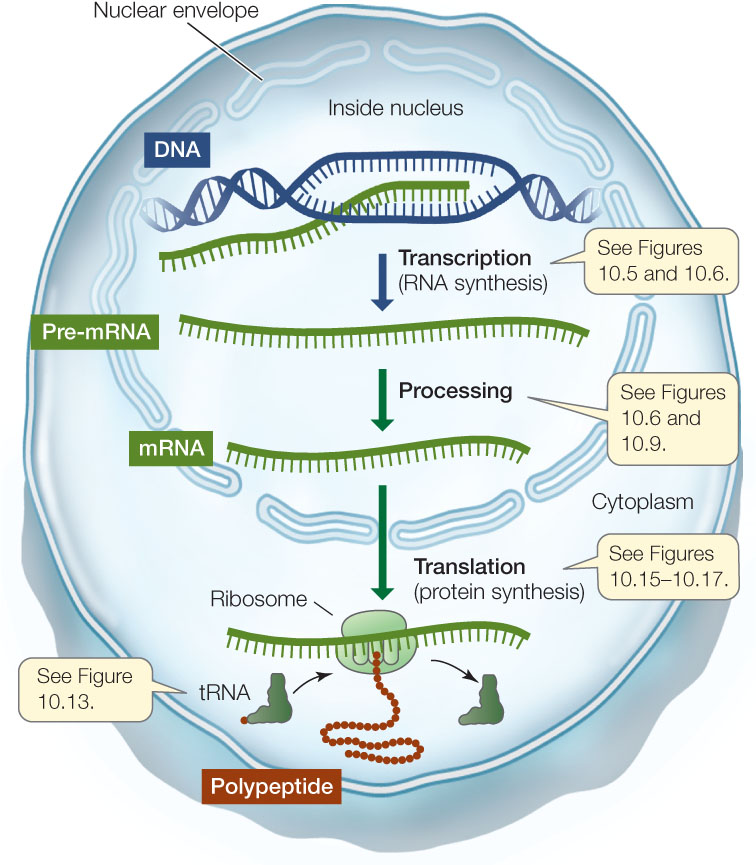 Figure 10.3: From Gene to Protein This diagram summarizes the processes of gene expression in eukaryotes.
Figure 10.3: From Gene to Protein This diagram summarizes the processes of gene expression in eukaryotes. - Ribosomal RNA and translation: The ribosome is essentially a protein synthesis factory with multiple proteins and several ribosomal RNAs (rRNAs). One of the rRNAs catalyzes peptide bond formation between amino acids, to form a polypeptide.
- Transfer RNA mediates between mRNA and protein: A third kind of RNA called transfer RNA (tRNA) can both bind a specific amino acid and recognize a specific sequence of nucleotides in mRNA, by complementary base pairing (A with U, and G with C). It is the tRNA that recognizes which amino acid should be added next to a growing poly-peptide chain (see Figure 10.3).
In Chapter 11 we will consider other RNAs, which play roles in the regulation of gene expression.
CHECKpointCONCEPT10.1
- What is the difference between the “one gene-one protein” and “one gene-one polypeptide” hypotheses?
- What is the difference between gene transcription and translation?
- Could a person inherit homozygous recessive alleles for both alkaptonuria and phenylketonuria? If so, what would the symptoms be?
- Defining phenotype as the presence of a polypeptide chain of a particular amino acid sequence, would you expect the Zürich variant of β-globin (see Figure 10.2) to be inherited as a dominant, recessive, or codominant? Explain your answer.
In this section we have shown how the connection between genes and phenotypes can be understood in terms of DNA and proteins. We will now turn to some details of the process of gene expression, which is at the heart of what genes do.