17.3Entry to the Citric Acid Cycle and Metabolism Through It Are Controlled
Entry to the Citric Acid Cycle and Metabolism Through It Are Controlled
The citric acid cycle is the final common pathway for the aerobic oxidation of fuel molecules. Moreover, as we will see shortly (Section 17.4) and repeatedly elsewhere in our study of biochemistry, the cycle is an important source of building blocks for a host of important biomolecules. As befits its role as the metabolic hub of the cell, entry into the cycle and the rate of the cycle itself are controlled at several stages.
511
The pyruvate dehydrogenase complex is regulated allosterically and by reversible phosphorylation
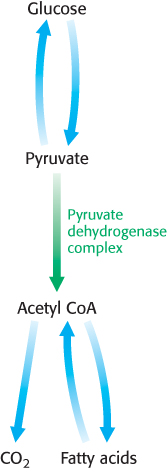
As stated earlier, glucose can be formed from pyruvate (Section 16.3). However, the formation of acetyl CoA from pyruvate is an irreversible step in animals and thus they are unable to convert acetyl CoA back into glucose. The oxidative decarboxylation of pyruvate to acetyl CoA commits the carbon atoms of glucose to one of two principal fates: oxidation to CO2 by the citric acid cycle, with the concomitant generation of energy, or incorporation into lipid (Figure 17.16). As expected of an enzyme at a critical branch point in metabolism, the activity of the pyruvate dehydrogenase complex is stringently controlled. High concentrations of reaction products inhibit the reaction: acetyl CoA inhibits the transacetylase component (E2) by binding directly, whereas NADH inhibits the dihydrolipoyl dehydrogenase (E3). High concentrations of NADH and acetyl CoA inform the enzyme that the energy needs of the cell have been met or that fatty acids are being degraded to produce acetyl CoA and NADH. In either case, there is no need to metabolize pyruvate to acetyl CoA. This inhibition has the effect of sparing glucose, because most pyruvate is derived from glucose by glycolysis (Section 16.1).
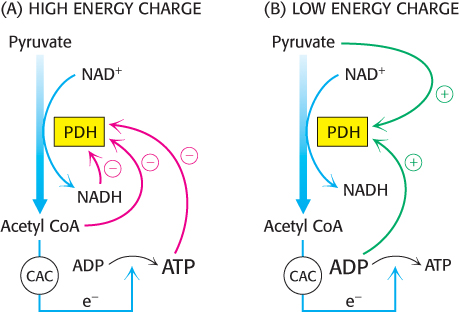
The key means of regulation of the complex in eukaryotes is covalent modification (Figure 17.17). Phosphorylation of the pyruvate dehydrogenase component (E1) by pyruvate dehydrogenase kinase (PDK) switches off the activity of the complex. There are four isozymes of PDK that are expressed in a tissue-
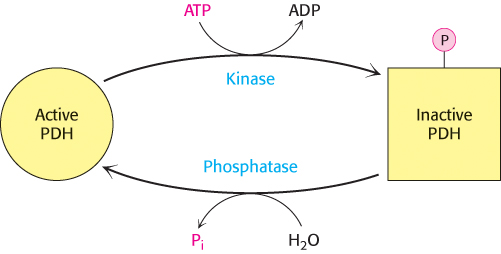
512
As exercise begins, the concentrations of ADP and pyruvate will increase as muscle contraction consumes ATP and glucose is converted into pyruvate to meet the energy demands. Both ADP and pyruvate activate the dehydrogenase by inhibiting the kinase. Moreover, the phosphatase is stimulated by Ca2+, the same signal that initiates muscle contraction. A rise in the cytoplasmic Ca2+ level (Section 35.2) elevates the mitochondrial Ca2+ level. The rise in mitochondrial Ca2+ activates the phosphatase, enhancing pyruvate dehydrogenase activity.
In some tissues, the phosphatase is regulated by hormones. In liver, epinephrine binds to the α-adrenergic receptor to initiate the phosphatidylinositol pathway (Section 14.1), causing an increase in Ca2+ concentration that activates the phosphatase. In tissues capable of fatty acid synthesis, such as the liver and adipose tissue, insulin, the hormone that signifies the fed state, stimulates the phosphatase, increasing the conversion of pyruvate into acetyl CoA. Acetyl CoA is the precursor for fatty acid synthesis (Section 22.4). In these tissues, the pyruvate dehydrogenase complex is activated to funnel glucose to pyruvate and then to acetyl CoA and ultimately to fatty acids.
 In people with a phosphatase deficiency, pyruvate dehydrogenase is always phosphorylated and thus inactive. Consequently, glucose is processed to lactate rather than acetyl CoA. This condition results in unremitting lactic acidosis—
In people with a phosphatase deficiency, pyruvate dehydrogenase is always phosphorylated and thus inactive. Consequently, glucose is processed to lactate rather than acetyl CoA. This condition results in unremitting lactic acidosis—
The citric acid cycle is controlled at several points
The rate of the citric acid cycle is precisely adjusted to meet an animal cell’s needs for ATP (Figure 17.19). The primary control points are the allosteric enzymes isocitrate dehydrogenase and α-ketoglutarate dehydrogenase, the first two enzymes in the cycle to generate high-
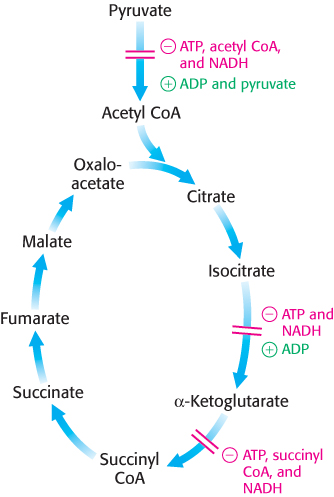
The first control site is isocitrate dehydrogenase. The enzyme is allosterically stimulated by ADP, which enhances the enzyme’s affinity for substrates. The binding of isocitrate, NAD+, Mg2+, and ADP is mutually cooperative. In contrast, ATP is inhibitory. The reaction product NADH also inhibits isocitrate dehydrogenase by directly displacing NAD+. It is important to note that several steps in the cycle require NAD+ or FAD, which are abundant only when the energy charge is low.
A second control site in the citric acid cycle is α-ketoglutarate dehydrogenase, which catalyzes the rate-
The use of isocitrate dehydrogenase and α-ketoglutarate dehydrogenase as control points integrates the citric acid cycle with other pathways and highlights the central role of the citric acid cycle in metabolism. For instance, the inhibition of isocitrate dehydrogenase leads to a buildup of citrate, because the interconversion of isocitrate and citrate is readily reversible under intracellular conditions. Citrate can be transported to the cytoplasm, where it signals phosphofructokinase to halt glycolysis (Section 16.2) and where it can serve as a source of acetyl CoA for fatty acid synthesis (Section 22.4). The α-ketoglutarate that accumulates when α-ketoglutarate dehydrogenase is inhibited can be used as a precursor for several amino acids and the purine bases (Chapter 23 and Chapter 25).
513
In many bacteria, the funneling of two-
Defects in the citric acid cycle contribute to the development of cancer
Four enzymes crucial to cellular respiration are known to contribute to the development of cancer: succinate dehydrogenase, fumarase, pyruvate dehydrogenase kinase and isocitrate dehydrogenase. Mutations that alter the activity of the first three of these enzymes enhance aerobic glycolysis (Section 16.2). In aerobic glycolysis, cancer cells preferentially metabolize glucose to lactate even in the presence of oxygen. Defects in these enzymes share a common biochemical link: the transcription factor hypoxia inducible factor 1 (HIF-
Normally, HIF-
Recent research suggestions that defects in the enzymes of the citric acid cycle may significantly affect the regulation of prolyl hydroxylase 2. When either succinate dehydrogenase or fumarase is defective, succinate and fumarate accumulate in the mitochondria and spill over into the cytoplasm. Both succinate and fumarate are competitive inhibitors of prolyl hydroxylase 2. The inhibition of prolyl hydroxylase 2 results in the stabilization of HIF-
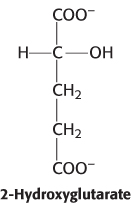
Mutations in isocitrate dehydrogenase result in the generation of an oncogenic metabolite, 2-
These observations linking citric acid cycle enzymes to cancer suggest that cancer is also a metabolic disease, not simply a disease of mutant growth factors and cell cycle control proteins. The realization that there is a metabolic component to cancer opens the door to new thinking about the control of cancer. Indeed, preliminary experiments suggest that if cancer cells undergoing aerobic glycolysis are forced by pharmacological manipulation to use oxidative phosphorylation, the cancer cells lose their malignant properties. It is also interesting to note that the citric acid cycle, which has been studied for decades, still has secrets to be revealed by future biochemists.
514