32.3
The Control of Gene Expression Can Require Chromatin Remodeling
Early observations suggested that chromatin structure plays a major role in controlling eukaryotic gene expression. DNA that is densely packaged into chromatin is less susceptible to cleavage by the nonspecific DNA-cleaving enzyme DNase I. Regions adjacent to genes that are being transcribed are more sensitive to cleavage by DNase I than are other sites in the genome, suggesting that the DNA in these regions is less compacted than it is elsewhere and more accessible to proteins. In addition, some sites, usually within 1 kb of the start site of an active gene, are exquisitely sensitive to DNase I and other nucleases. These hypersensitive sites correspond to regions that have few nucleosomes or contain nucleosomes in an altered conformation Hypersensitive sites are cell-type specific and developmentally regulated. For example, globin genes in the precursors of erythroid cells from 20-hour-old chicken embryos are insensitive to DNase I. However, when hemoglobin synthesis begins at 35 hours, regions adjacent to these genes become highly susceptible to digestion. In tissues such as the brain that produce no hemoglobin, the globin genes remain resistant to DNase I throughout development and into adulthood. These studies suggest that a prerequisite for gene expression is a relaxing of the chromatin structure.
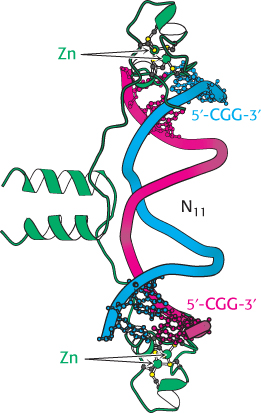
 FIGURE 32.13 GAL4 binding sites. The yeast transcription factor GAL4 binds to DNA sequences of the form 5′-CGG(N)11CCG-3′. Two zinc-based domains are present in the DNA-binding region of this protein. Notice that these domains contact the 5′-CGG-35′ sequences, leaving the center of the site uncontacted.
FIGURE 32.13 GAL4 binding sites. The yeast transcription factor GAL4 binds to DNA sequences of the form 5′-CGG(N)11CCG-3′. Two zinc-based domains are present in the DNA-binding region of this protein. Notice that these domains contact the 5′-CGG-35′ sequences, leaving the center of the site uncontacted.
[Drawn from 1D66.pdb.]
Recent experiments even more clearly revealed the role of chromatin structure in regulating access to DNA-binding sites. Genes required for galactose utilization in yeast are activated by a transcription factor called GAL4, which recognizes DNA-binding sites with two 5′-CGG-3′ sequences on complementary strands separated by 11 base pairs (Figure 32.13). Approximately 4000 potential GAL4 binding sites of the form 5′-CGG(N)11CCG-3′ are present in the yeast genome, but only 10 of them regulate genes necessary for galactose metabolism. How is GAL4 targeted to only a small fraction of the potential binding sites? This question is addressed through the use of a technique called chromatin immunoprecipitation (ChIP; Figure 32.14). GAL4 is first cross-linked to its DNA-binding sites in chromatin. The DNA is then fragmented into small pieces, and antibodies to GAL4 are used to isolate the chromatin fragments containing GAL4. The cross-linking is reversed, and the DNA is isolated and characterized. The results of these studies reveal that only approximately 10 of the 4000 potential GAL4 sites are occupied by GAL4 when the cells are growing on galactose; more than 99% of the sites appear to be blocked, presumably by the local chromatin structure. Thus, whereas in prokaryotes all sites appear to be equally accessible, chromatin structure shields a large number of the potential binding sites in eukaryotic cells. GAL4 is thereby prevented from binding to sites that are unimportant in galactose metabolism. These lines of evidence and others reveal that chromatin structure is altered in active genes compared with inactive ones.
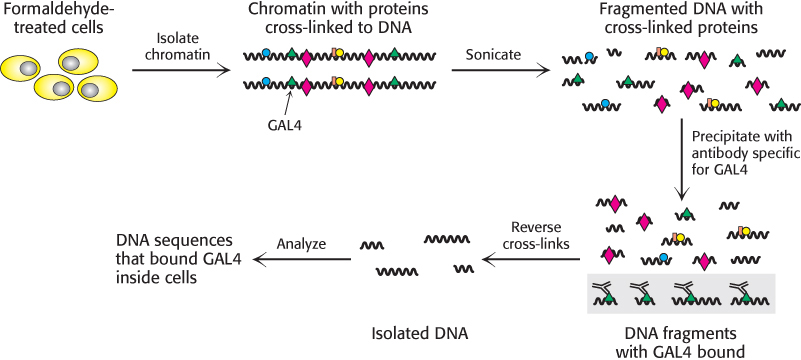
FIGURE 32.14Chromatin immunoprecipitation. Cells or isolated nuclei are treated with formaldehyde to cross-link proteins to DNA. The cells are then lysed and the DNA is fragmented by sonication. DNA fragments bound to a particular protein are isolated through the use of an antibody specific for that protein. The cross-links are then reversed and the DNA fragments are characterized.
The methylation of DNA can alter patterns of gene expression
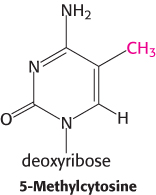
The degree of methylation of DNA provides another mechanism, in addition to packaging with histones, for inhibiting gene expression inappropriate to a specific cell type. Carbon 5 of cytosine can be methylated by specific methyltransferases. About 70% of the 5′-CpG-3′ sequences (where “p” represents the phosphate residue in the DNA backbone) in mammalian genomes are methylated. However, the distribution of these methylated cytosines varies, depending on the cell type. Consider the β-globin gene. In cells that are actively expressing hemoglobin, the region from approximately 1 kb upstream of the start site to approximately 100 bp downstream of the start site is less methylated than the corresponding region in cells that do not express this gene. The relative absence of 5-methylcytosines near the start site is referred to as hypomethylation. The methyl group of 5-methylcytosine protrudes into the major groove where it could easily interfere with the binding of proteins that stimulate transcription.
 The distribution of CpG sequences in mammalian genomes is not uniform. Many CpG sequences have been converted into TpG through mutation by the deamination of 5-methylcytosine to thymine. However, sites near the 5′ ends of genes have been maintained because of their role in gene expression. Thus, most genes are found in CpG islands, regions of the genome that contain approximately four times as many CpG sequences as does the remainder of the genome.
The distribution of CpG sequences in mammalian genomes is not uniform. Many CpG sequences have been converted into TpG through mutation by the deamination of 5-methylcytosine to thymine. However, sites near the 5′ ends of genes have been maintained because of their role in gene expression. Thus, most genes are found in CpG islands, regions of the genome that contain approximately four times as many CpG sequences as does the remainder of the genome.
Steroids and related hydrophobic molecules pass through membranes and bind to DNA-binding receptors
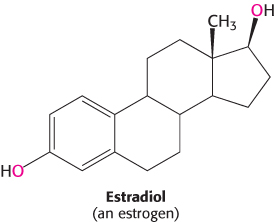
We next look at an example that illustrates how transcription factors can stimulate changes in chromatin structure that affect transcription. We will consider in some detail the system that detects and responds to estrogens. Synthesized and released by the ovaries, estrogens, such as estradiol, are cholesterol-derived steroid hormones (Section 26.4). They are required for the development of female secondary sex characteristics and, along with progesterone, participate in the ovarian cycle.
Because they are hydrophobic molecules, estrogens easily diffuse across cell membranes. When inside a cell, estrogens bind to highly specific, soluble receptor proteins. Estrogen receptors are members of a large family of proteins that act as receptors for a wide range of hydrophobic molecules, including other steroid hormones, thyroid hormones, and retinoids.
The human genome encodes approximately 50 members of this family, often referred to as nuclear hormone receptors. The genomes of other multicellular eukaryotes encode similar numbers of nuclear hormone receptors, although they are absent in yeast.
All these receptors have a similar mode of action. On binding of the signal molecule (called, generically, a ligand), the ligand–receptor complex modifies the expression of specific genes by binding to control elements in the DNA. Estrogen receptors bind to specific DNA sites (referred to as estrogen response elements or EREs) that contain the consensus sequence 5′-AGGTCANNNTGACCT-3′. As expected from the symmetry of this sequence, an estrogen receptor binds to such sites as a dimer.
A comparison of the amino acid sequences of members of this family reveals two highly conserved domains: a DNA-binding domain and a ligand-binding domain (Figure 32.15). The DNA-binding domain lies toward the center of the molecule and consists of a set of zinc-based domains different from the Cys2His2 zinc-finger proteins introduced in Section 32.2. These zinc-based domains bind to specific DNA sequences by virtue of an α helix that lies in the major groove in the specific DNA complexes formed by estrogen receptors.
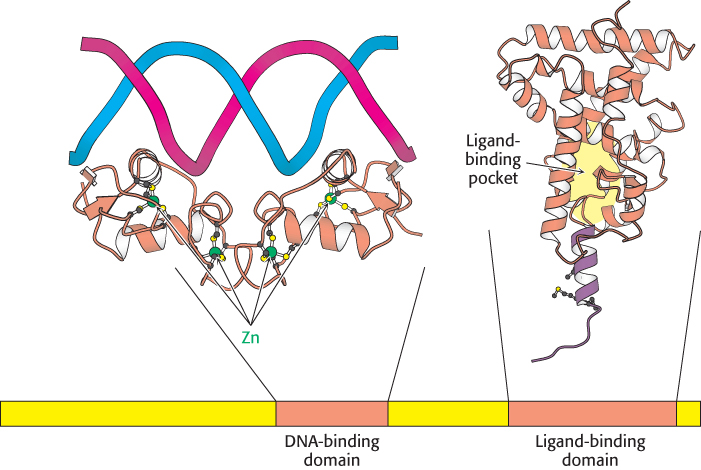
FIGURE 32.15 Structure of two nuclear hormone-receptor domains. Nuclear hormone receptors contain two crucial conserved domains: (1) a DNA-binding domain toward the center of the sequence and (2) a ligand-binding domain toward the carboxyl terminus. The structure of a dimer of the DNA-binding domain bound to DNA is shown, as is one monomer of the normally dimeric ligand-binding domain.
[Drawn from 1HCQ and 1LBD.pdb.]
Nuclear hormone receptors regulate transcription by recruiting coactivators to the transcription complex
The second highly conserved domain of the nuclear receptor proteins lies near the carboxyl terminus and is the ligand-binding site. This domain folds into a structure that consists almost entirely of α helices, arranged in three layers. The ligand binds in a hydrophobic pocket that lies in the center of this array of helices (Figure 32.16). This domain changes conformation when it binds its ligand, estrogen. How does ligand binding lead to changes in gene expression? The simplest model would have the binding of ligand alter the DNA-binding properties of the receptor, analogously to the lac repressor in prokaryotes. However, experiments with purified nuclear hormone receptors revealed that ligand binding does not significantly alter DNA-binding affinity and specificity. Another mechanism is operative.
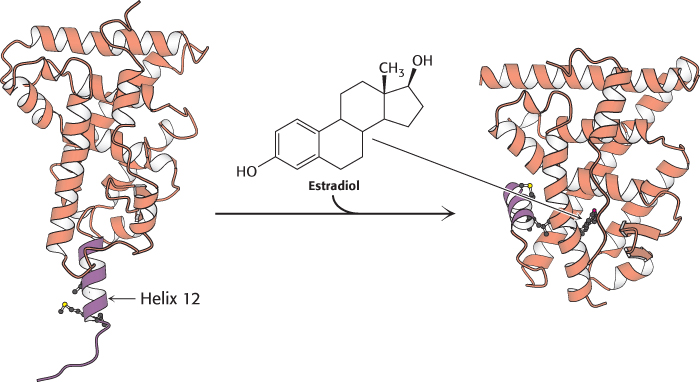
FIGURE 32.16 Ligand binding to nuclear hormone receptor. The ligand lies completely surrounded within a pocket in the ligand-binding domain. Notice that the last α helix, helix 12 (shown in purple), folds into a groove on the side of the structure on ligand binding.
[Drawn from 1LDB and 1ERE.pdb.]
Investigators sought to determine whether specific proteins might bind to the nuclear hormone receptors only in the presence of ligand. Such searches led to the identification of several related proteins called coactivators, such as SRC-1 (steroid receptor coactivator-1), GRIP-1 (glucocorticoid receptor interacting protein-1), and NcoA-1 (nuclear hormone receptor coactivator-1). These coactivators are referred to as the p160 family because of their size. The binding of ligand to the receptor induces a conformational change that allows the recruitment of a coactivator (Figure 32.17). In many cases, these coactivators are enzymes that catalyze reactions that lead to the modification of chromatin structure.
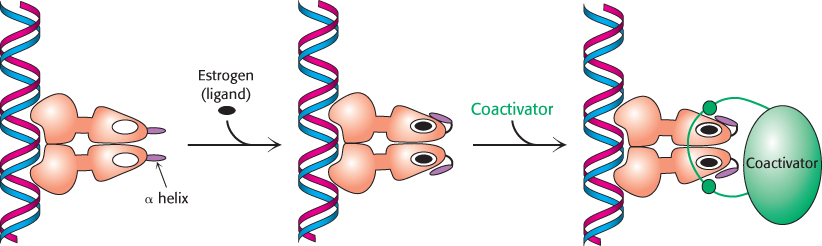
FIGURE 32.17Coactivator recruitment. The binding of ligand to a nuclear hormone receptor induces a conformational change in the ligand-binding domain. This change in conformation generates favorable sites for the binding of a coactivator.
Steroid-hormone receptors are targets for drugs
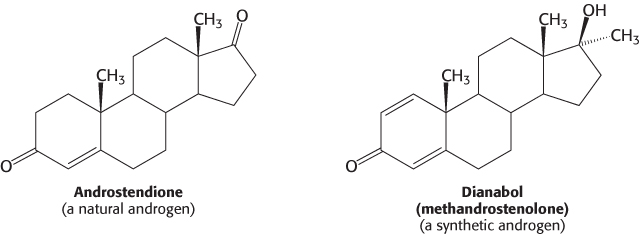
 Molecules such as estradiol that bind to a receptor and trigger signaling pathways are called agonists. Athletes sometimes take natural and synthetic agonists of the androgen receptor, a member of the family of nuclear hormone receptors, because their binding to the androgen receptor stimulates the expression of genes that enhance the development of lean muscle mass.
Molecules such as estradiol that bind to a receptor and trigger signaling pathways are called agonists. Athletes sometimes take natural and synthetic agonists of the androgen receptor, a member of the family of nuclear hormone receptors, because their binding to the androgen receptor stimulates the expression of genes that enhance the development of lean muscle mass.
Referred to as anabolic steroids, such compounds used in excess are not without side effects. In men, excessive use leads to a decrease in the secretion of testosterone, to testicular atrophy, and sometimes to breast enlargement (gynecomastia) if some of the excess androgen is converted into estrogen. In women, excess testosterone causes a decrease in ovulation and estrogen secretion; it also causes breast regression and growth of facial hair.
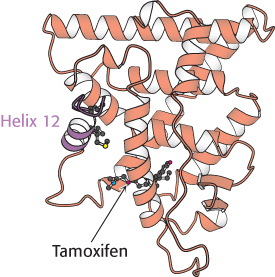
FIGURE 32.18 Estrogen receptor—tamoxifen complex. Tamoxifen binds in the pocket normally occupied by estrogen. However, notice that part of the tamoxifen structure extends from this pocket, and so helix 12 cannot pack in its usual position. Instead, this helix blocks the coactivator-binding site.
[Drawn from 3ERT.pdb.]
Other molecules bind to nuclear hormone receptors but do not effectively trigger signaling pathways. Such compounds are called antagonists and are, in many ways, like competitive inhibitors of enzymes. Some important drugs are antagonists that target the estrogen receptor. For example, tamoxifen and raloxifene are used in the treatment and prevention of breast cancer, because some breast tumors rely on estrogen-mediated pathways for growth. Because some of these compounds have distinct effects on different forms of the estrogen receptor, they are referred to as selective estrogen receptor modulators (SERMs).
The determination of the structures of complexes between the estrogen receptor and these drugs revealed the basis for their antagonist effect (Figure 32.18). Tamoxifen binds to the same site as estradiol does. However, tamoxifen has a group that extends out of the normal ligand-binding pocket, as do other antagonists. These groups block the normal conformational changes induced by estrogen. Tamoxifen blocks the binding of coactivators and thus inhibits the activation of gene expression.
Chromatin structure is modulated through covalent modifications of histone tails
We have seen that nuclear receptors respond to signal molecules by recruiting coactivators. Now we can ask, How do coactivators modulate transcriptional activity? These proteins act to loosen the histone complex from the DNA, exposing additional DNA regions to the transcription machinery.
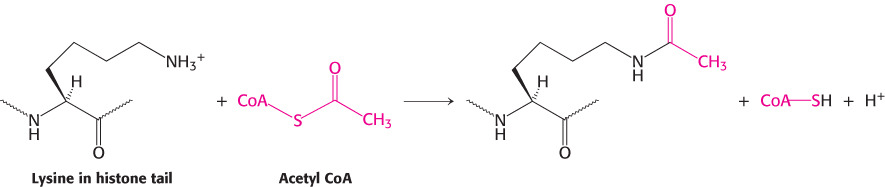
Much of the effectiveness of coactivators appears to result from their ability to covalently modify the amino-terminal tails of histones as well as regions on other proteins. Some of the p160 coactivators and the proteins that they recruit catalyze the transfer of acetyl groups from acetyl CoA to specific lysine residues in these amino-terminal tails.
Enzymes that catalyze such reactions are called histone acetyltransferases (HATs). The histone tails are readily extended; so they can fit into the HAT active site and become acetylated (Figure 32.19).
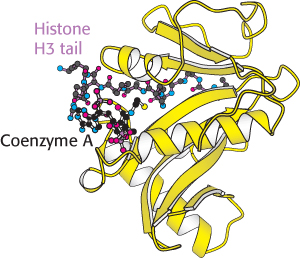
FIGURE 32.19 Structure of histone acetyltransferase. The amino-terminal tail of histone H3 extends into a pocket in which a lysine side chain can accept an acetyl group from acetyl Coenzyme A bound in an adjacent site.
[Drawn from 1QSN.pdb.]
What are the consequences of histone acetylation? Lysine bears a positively charged ammonium group at neutral pH. The addition of an acetyl group generates an uncharged amide group. This change dramatically reduces the affinity of the tail for DNA and modestly decreases the affinity of the entire histone complex for DNA, loosening the histone complex from the DNA.
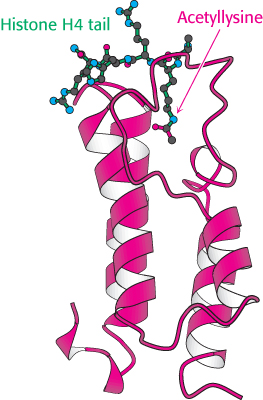
FIGURE 32.20 Structure of a bromodomain. This four-helix-bundle domain binds peptides containing acetyllysine. Notice that an acetylated peptide from histone H4 is bound in the structure.
[Drawn from 1EGI.pdb.]
In addition, the acetylated lysine residues interact with a specific acetyllysine-binding domain that is present in many proteins that regulate eukaryotic transcription. This domain, termed a bromodomain, comprises approximately 110 amino acids that form a four-helix bundle containing a peptide-binding site at one end (Figure 32.20).
Bromodomain-containing proteins are components of two large complexes essential for transcription. One is a complex of more than 10 polypeptides that binds to the TATA-box-binding protein. Recall that the TATA-box-binding protein is an essential transcription factor for many genes (Section 29.2). Proteins that bind to the TATA-box-binding protein are called TAFs (for TATA-box-binding protein associated factors). In particular, TAF1 contains a pair of bromodomains near its carboxyl terminus. The two domains are oriented such that each can bind one of two acetyllysine residues at positions 5 and 12 in the histone H4 tail. Thus, acetylation of the histone tails provides a mechanism for recruiting other components of the transcriptional machinery.
Bromodomains are also present in some components of large complexes known as chromatin-remodeling complexes or chromatin-remodeling engines. These complexes, which also contain domains homologous to those of helicases, utilize the free energy of ATP hydrolysis to shift the positions of nucleosomes along the DNA and to induce other conformational changes in chromatin (Figure 32.21). Histone acetylation can lead to a reorganization of the chromatin structure, potentially exposing binding sites for other factors. Thus, histone acetylation can activate transcription through a combination of three mechanisms: by reducing the affinity of the histones for DNA, by recruiting other components of the transcriptional machinery, and by initiating the remodeling of the chromatin structure.
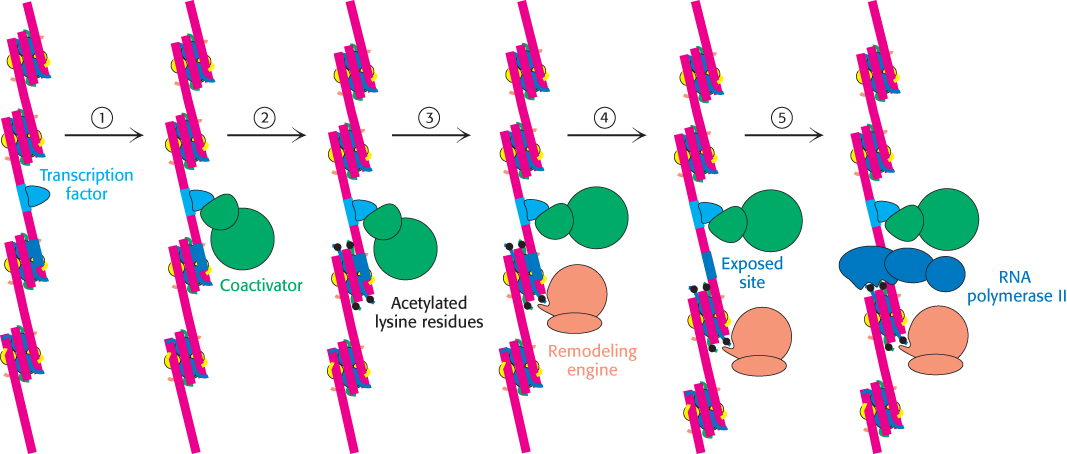
FIGURE 32.21Chromatin remodeling. Eukaryotic gene regulation begins with an activated transcription factor bound to a specific site on DNA. One scheme for the initiation of transcription by RNA polymerase II requires five steps: (1) recruitment of a coactivator, (2) acetylation of lysine residues in the histone tails, (3) binding of a remodeling-engine complex to the acetylated lysine residues, (4) ATP-dependent remodeling of the chromatin structure to expose a binding site for RNA polymerase II or for other factors, and (5) recruitment of RNA polymerase II. Only two subunits are shown for each complex, although the actual complexes are much larger. Other schemes are possible.
Nuclear hormone receptors also include regions that interact with components of the mediator complex. Thus, two mechanisms of gene regulation can work in concert. Modification of histones and chromatin remodeling can open up regions of chromatin into which the transcription complex can be recruited through protein–protein interactions.
Histone deacetylases contribute to transcriptional repression
Just as in prokaryotes, some changes in a cell’s environment lead to the repression of genes that had been active. The modification of histone tails again plays an important role. However, in repression, a key reaction appears to be the deacetylation of acetylated lysine, catalyzed by specific histone deacetylase enzymes.
TABLE 32.2 Selected histone modifications
In many ways, the acetylation and deacetylation of lysine residues in histone tails (and, likely, in other proteins) is analogous to the phosphorylation and dephosphorylation of serine, threonine, and tyrosine residues in other stages of signaling processes. Like the addition of phosphoryl groups, the addition of acetyl groups can induce conformational changes and generate novel binding sites. Without a means of removal of these groups, however, these signaling switches will become stuck in one position and lose their effectiveness. Like phosphatases, deacetylases help reset the switches.
Acetylation is not the only modification of histones and other proteins in gene-regulation processes. The methylation of specific lysine and arginine residues also can be important. Some of the more common modifications are shown in Table 32.2.
The elucidation of the roles of these modifications is a very active area of research at present. The relation between various histone modifications and their roles in controlling gene expression is sometimes referred to as “the histone code.” Although important generalizations have been discovered, this code clearly is subtle and complicated rather than being a set of hard and fast rules. Nonetheless, the development and application of methods that allow probing of histone modifications on a genome-wide scale in a range of cell types has provided large data sets that can be examined to test and refine models.