Serendipitous observations can drive drug development
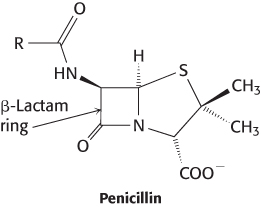
Perhaps the most well-known observation in the history of drug development is Alexander Fleming’s chance observation in 1928 that colonies of the bacterium Staphylococcus aureus died when they were adjacent to colonies of the mold Penicillium notatum. Spores of the mold had landed accidentally on plates growing the bacteria. Fleming soon realized that the mold produced a substance that could kill disease-causing bacteria. This discovery led to a fundamentally new approach to the treatment of bacterial infections. Howard Florey and Ernest Chain developed a powdered form of the substance, termed penicillin, that became a widely used antibiotic in the 1940s. When the structure of penicillin was elucidated in 1945, it was found to contain a four-membered β-lactam ring. This unusual feature is key to the antibacterial function of penicillin, as noted earlier (Section 8.5).
Three steps were crucial to fully capitalize on Fleming’s discovery. First, an industrial process was developed for the production of penicillin from Penicillium mold on a large scale. Second, penicillin and its derivatives were chemically synthesized. The availability of synthetic penicillin derivatives opened the way for scientists to explore the relations between structure and function. Many such penicillin derivatives have found widespread use in medicine. Finally, in 1965, Jack Strominger and James Park independently determined that penicillin exerts its antibiotic activity by blocking a critical transpeptidase reaction in bacterial cell-wall biosynthesis (Figure 36.15), as introduced in Section 8.5.
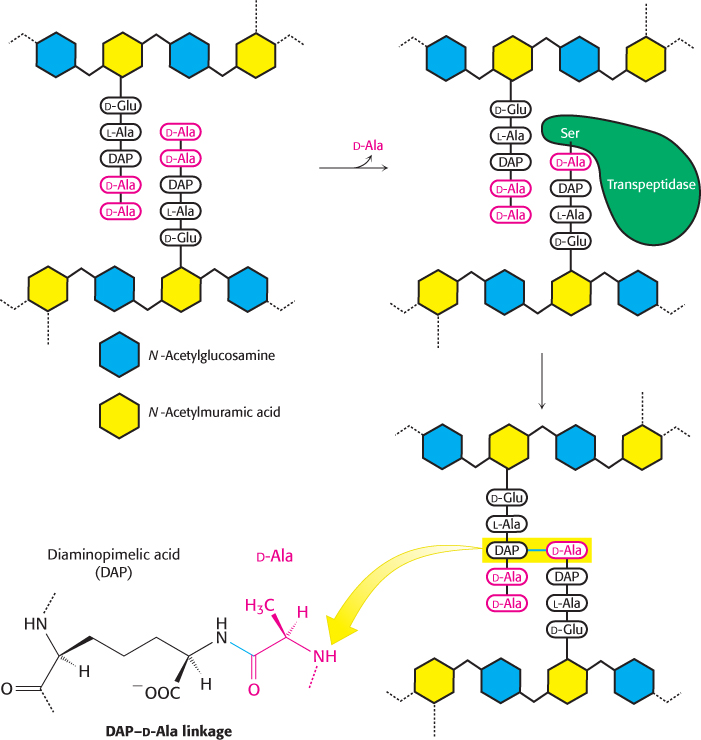
FIGURE 36.15Mechanism of cell-wall biosynthesis disrupted by penicillin. A transpeptidase enzyme catalyzes the formation of cross-links between peptidoglycan groups. In the case shown, the transpeptidase catalyzes the linkage of d-alanine at the end of one peptide chain to the amino acid diaminopimelic acid (DAP) on another peptide chain. The diaminopimelic acid linkage (bottom left) is found in Gram-negative bacteria such as E. coli. Linkages of glycine-rich peptides are found in Gram-positive bacteria. Penicillin inhibits the action of the transpeptidase; so bacteria exposed to the drug have weak cell walls that are susceptible to lysis.
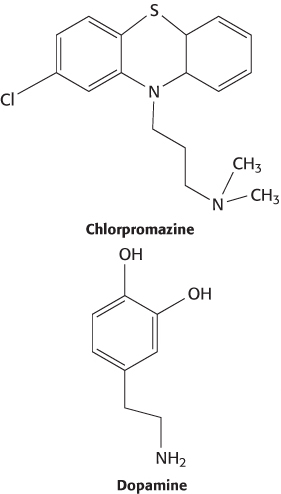
Many other drugs have been discovered by serendipitous observations. Chlorpromazine (Thorazine), a drug used to treat psychosis, was discovered in the course of investigations directed toward the treatment of shock in surgical patients. In 1952, French surgeon Henri Laborit noticed that, after taking the compound, his patients were remarkably calm. This observation suggested that chlorpromazine could benefit psychiatric patients, and, indeed, the drug has been used for many years to treat patients with schizophrenia and bipolar disorder. The drug does have significant side effects, and its use has been largely superseded by more recently developed drugs.
Chlorpromazine acts by binding to receptors for the neurotransmitter dopamine and blocking them. Dopamine D2 receptors are the targets of many other psychoactive drugs. In the search for drugs with more-limited side effects, studies have been performed to correlate drug effects with biochemical parameters such as dissociation constants and binding and release rate constants.
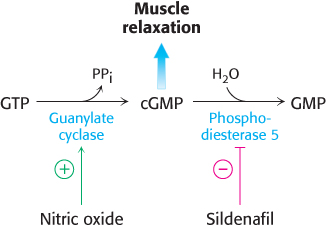
FIGURE 36.17Muscle-relaxation pathway. Increases in nitric oxide levels stimulate guanylate cyclase, which produces cGMP. The increased cGMP concentration promotes smooth-muscle relaxation. PDE5 hydrolyzes cGMP, which lowers the cGMP concentration. The inhibition of PDE5 by sildenafil maintains elevated levels of cGMP.
A more recent example of a drug discovered by chance observation is sildenafil (Viagra). This compound was developed as an inhibitor of phosphodiesterase 5 (PDE5), an enzyme that catalyzes the hydrolysis of cGMP to GMP (Figure 36.16). The compound was intended as a treatment for hypertension and angina because cGMP plays a central role in the relaxation of smooth-muscle cells in blood vessels (Figure 36.17). The inhibition of PDE5 was expected to increase the concentration of cGMP by blocking the pathway for its degradation. In the course of early clinical trials in Wales, some men reported unusual penile erections. Whether this chance observation by a few men was due to the compound or to other effects was unclear. However, the observation made some biochemical sense because smooth-muscle relaxation due to increased cGMP levels had been discovered to play a role in penile erection. Subsequent clinical trials directed toward the evaluation of sildenafil for erectile dysfunction were successful. This account testifies to the importance of collecting comprehensive information from clinical-trial participants. In this case, incidental observations led to a new treatment for erectile dysfunction and a multibillion-dollar-per-year drug market.
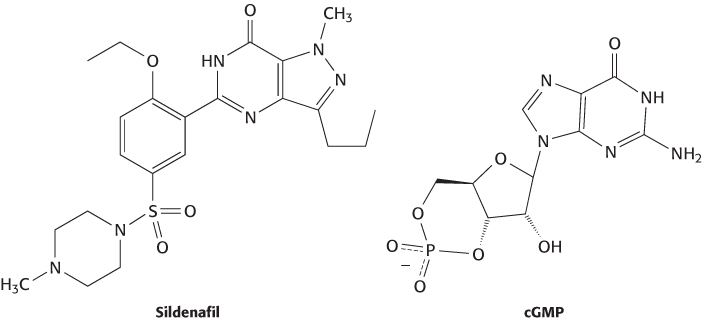
FIGURE 36.16Sildenafil, a mimic of cGMP. Sildenafil was designed to resemble cGMP, the substrate of phosphodiesterase 5 (PDE5).
Natural products are a valuable source of drugs and drug leads
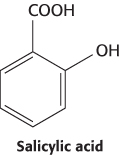
No drug is as widely used as aspirin. Observers at least as far back as Hippocrates (~400 b.c.) have noted the use of extracts from the bark and leaves of the willow tree for pain relief. In 1829, a mixture called salicin was isolated from willow bark. Subsequent analysis identified salicylic acid as the active component of this mixture. Salicylic acid was formerly used to treat pain, but this compound often irritated the stomach. Several investigators attempted to find a means to neutralize salicylic acid. Felix Hoffmann, a chemist working at the German company Bayer, developed a less-irritating derivative by treating salicylic acid with a base and acetyl chloride. This derivative, acetylsalicylic acid, was named aspirin from “a” for acetyl chloride, “spir” for Spiraea ulmaria (meadowsweet, a flowering plant that also contains salicylic acid), and “in” (a common ending for drugs). Each year, approximately 35,000 tons of aspirin are taken worldwide, nearly the weight of the Titanic.
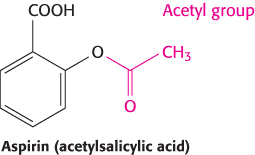
As discussed in Chapter 12, the acetyl group in aspirin is transferred to the side chain of a serine residue that lies along the path to the active site of the cyclooxygenase component of prostaglandin H2 synthase (Figure 12.24). In this position, the acetyl group blocks access to the active site. Thus, even though aspirin binds in the same pocket on the enzyme as salicylic acid, the acetyl group of aspirin dramatically increases its effectiveness as a drug. This account illustrates the value of screening extracts from plants and other materials that are believed to have medicinal properties for active compounds. The large number of herbal and folk medicines are a treasure trove of new drug leads.
Let us consider another example, which has also had a significant impact on the clinical management of individuals with cardiovascular disease. More than 100 years ago, a fatty, yellowish material was discovered on the arterial walls of patients who had died of vascular disease. The presence of the material was termed atheroma from the Greek word for porridge. This material proved to be cholesterol. The Framingham heart study, initiated in 1948, documented a correlation between high blood-cholesterol levels and high mortality rates from heart disease. This observation led to the notion that blocking cholesterol synthesis might lower blood-cholesterol levels and, in turn, lower the risk of heart disease. Initial attempts at blocking cholesterol synthesis focused on steps near the end of the pathway. However, these efforts were abandoned because the accumulation of the insoluble substrate for the inhibited enzyme led to the development of cataracts and other side effects. Investigators eventually identified a more-favorable target—namely, the enzyme HMG-CoA reductase (Section 26.2). This enzyme acts on a substrate, HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A), that does not accumulate, because it is water-soluble and can be utilized by other pathways.
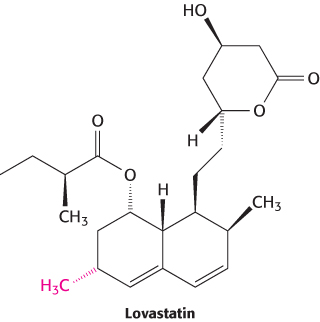
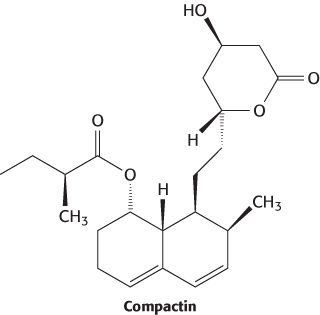
A promising natural product, compactin, was discovered in a screen of compounds from a fermentation broth from Penicillium citrinum in a search for antibacterial agents. In some, but not all, animal studies, compactin was found to inhibit HMG-CoA reductase and to lower serum cholesterol levels. In 1982, a new HMG-CoA reductase inhibitor was discovered in a fermentation broth from Aspergillus cereus. This compound, now called lovastatin, was found to be structurally very similar to compactin, bearing one additional methyl group.
In clinical trials, lovastatin significantly reduced serum cholesterol levels with few side effects. Most side effects could be prevented by treatment with mevalonate (the product of HMG-CoA reductase), indicating that the side effects were likely due to the highly effective blocking of HMG-CoA reductase. One notable side effect is muscle pain or weakness (termed myopathy), although its cause remains to be fully established. After many studies, the Food and Drug Administration (FDA) approved lovastatin for treating high serum cholesterol levels.
A structurally related HMG-CoA reductase inhibitor was later shown to cause a statistically significant decrease in deaths due to coronary heart disease. This result validated the benefits of lowering serum cholesterol levels. Further mechanistic analysis revealed that the HMG-CoA reductase inhibitor acts not only by lowering the rate of cholesterol biosynthesis, but also by inducing the expression of the low-density-lipoprotein (LDL) receptor (Section 26.3). Cells with such receptors remove LDL particles from the bloodstream, and so these particles cannot contribute to atheroma.
Screening libraries of synthetic compounds expands the opportunity for identification of drug leads
Lovastatin and its relatives are either natural products or readily derived from natural products. After the discovery of these compounds, totally synthetic molecules were developed that are more-potent inhibitors of HMG-CoA reductase (Figure 36.18). These compounds were found to be effective at lower dose levels, reducing side effects. The original HMG-CoA reductase inhibitors or their precursors were found by screening libraries of natural products. More recently, drug developers have tried screening large libraries of both natural products and purely synthetic compounds prepared in the course of many drug-development programs. Under favorable circumstances, hundreds of thousands or even millions of compounds can be tested in this process, termed high-throughput screening. Compounds in these libraries can be synthesized one at a time for testing. An alternative approach is to synthesize a large number of structurally related compounds that differ from one another at only one or a few positions all at once. This approach is often termed combinatorial chemistry. Here, compounds are synthesized with the use of the same chemical reactions but a variable set of reactants. Suppose that a molecular scaffold is constructed with two reactive sites and that 20 reactants can be used in the first site and 40 reactants can be used in the second site. A total of 20 × 40 = 800 possible compounds can be produced.
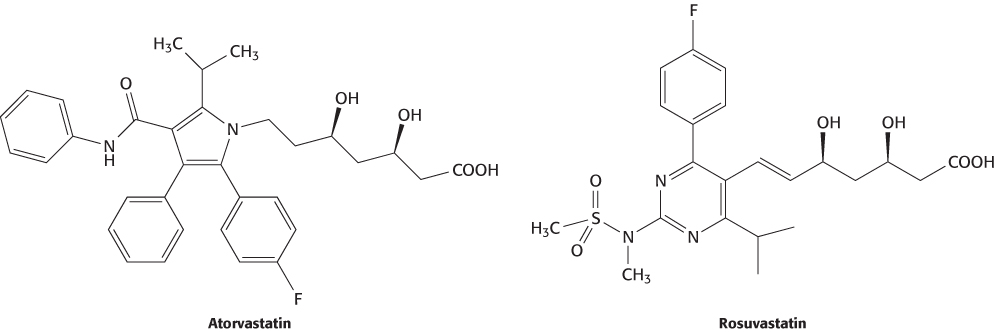
FIGURE 36.18Synthetic statins. Atorvastatin (Lipitor) and rosuvastatin (Crestor) are completely synthetic drugs that inhibit HMG-CoA reductase.
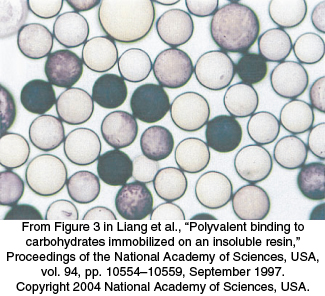
FIGURE 36.20Screening a library of synthesized carbohydrates. A small combinatorial library of carbohydrates synthesized on the surface of 130-mm beads is screened for carbohydrates that are bound tightly by a lectin from peanuts. Beads that have such carbohydrates are darkly stained through the action of an enzyme linked to the lectin.
A key method in combinatorial chemistry is split-pool synthesis (Figure 36.19). The method depends on solid-phase synthetic methods, first developed for the synthesis of peptides (Section 3.5). Compounds are synthesized on small beads. Beads containing an appropriate starting scaffold are produced and divided (split) into n sets, with n corresponding to the number of building blocks to be used at one site. Reactions adding the reactants at the first site are run, and the beads are isolated by filtration. The n sets of beads are then combined (pooled), mixed, and split again into m sets, with m corresponding to the number of reactants to be used at the second site. Reactions adding these m reactants are run, and the beads are again isolated. The important result is that each bead contains only one compound, even though the entire library of beads contains many. Furthermore, although only n + m reactions were run, n × m compounds are produced. With the preceding values for n and m, 20 + 40 = 60 reactions produce 20 × 40 = 800 compounds. In some cases, assays can be performed directly with the compounds still attached to the bead to find compounds with desired properties (Figure 36.20). Alternatively, each bead can be isolated and the compound can be cleaved from the bead to produce free compounds for analysis. After an interesting compound has been identified, analytical methods of various types must be used to identify which of the n × m compounds is present.
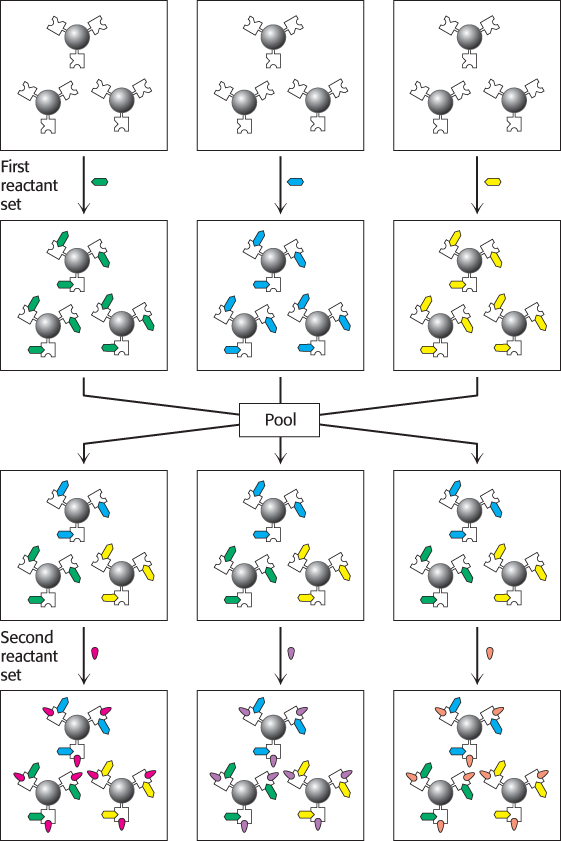
FIGURE 36.19Split-pool synthesis. Reactions are performed on beads. Each of the reactions with the first set of reactants is performed on a separate set of beads. The beads are then pooled, mixed, and split into sets. The second set of reactants is then added. Many different compounds will be produced, but all of the compounds on a single bead will be identical.
Note that the “universe” of druglike compounds is vast. More than an estimated 1040 compounds are possible with molecular weights less than 750. Thus, even with “large” libraries of millions of compounds, only a tiny fraction of the chemical possibilities are present for study.
Drugs can be designed on the basis of three-dimensional structural information about their targets
Many drugs bind to their targets in a manner reminiscent of Emil Fischer’s lock and key (Figure 8.8). Therefore, we should be able to design a key, given enough knowledge about the shape and chemical composition of the lock. In the idealized case, we would like to design a small molecule that is complementary in shape and electronic structure to a target protein so that it binds effectively to the targeted site. Despite our ability to determine three-dimensional structures rapidly, the achievement of this goal remains in the future. Designing stable compounds from scratch that have the correct shape and other properties to fit precisely into a binding site is difficult because predicting the structure that will best fit into a binding site is difficult. Prediction of binding affinity requires a detailed understanding of the interactions between a compound and its binding partner and of the interactions between the compound and the solvent when the compound is free in solution.
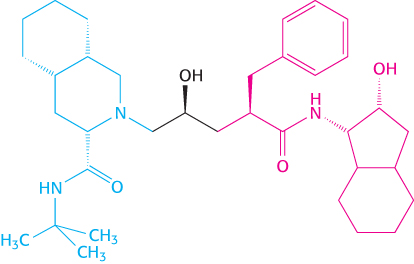
FIGURE 36.21Initial design of an HIV protease inhibitor. This compound was designed by combining part of one compound with good inhibition activity but poor solubility (shown in red) with part of another compound with better solubility (shown in blue).
Nonetheless, structure-based drug design has proved to be a powerful tool in drug development. Among its most prominent successes has been the development of drugs that inhibit the protease from the human immunodeficiency virus (HIV; Section 34.4). Consider the development of the protease inhibitor indinavir (Crixivan; Figure 9.19). Two sets of promising inhibitors that had high potency but poor solubility and bioavailability were discovered. X-ray crystallographic analysis and molecular-modeling findings suggested that a hybrid of these two inhibitor sets might have both high potency and improved bioavailability (Figure 36.21). The synthesized hybrid compound did show improvements but required further optimization. The structural data suggested one point where modifications could be tolerated. A series of compounds were prepared and tested for their ability to inhibit the protease (Figure 36.22). These data are a demonstration of a structure–activity relationship (SAR); they provide an opportunity to correlate structure with function and guide design of further molecules. The most-active compound showed poor bioavailability, but one of the other compounds (highlighted in yellow in Figure 36.22) showed good bioavailability and acceptable activity. The maximum serum concentration available through oral administration was significantly higher than the levels required to suppress replication of the virus. This drug, as well as other protease inhibitors developed at about the same time, has been used in combination with other drugs to treat AIDS with much more encouraging results than had been obtained previously (Figure 36.23).
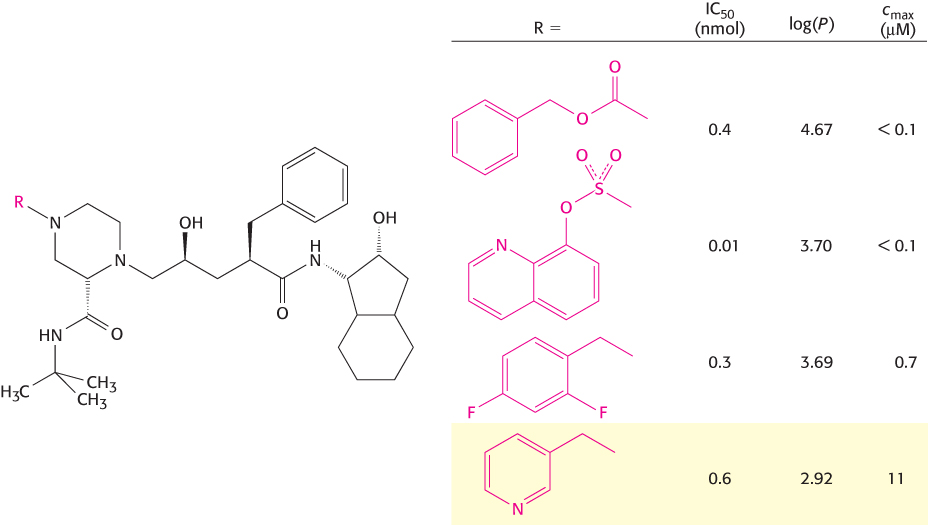
FIGURE 36.22Compound optimization. Four compounds are evaluated for characteristics including the IC50, log(P), and cmax (the maximal concentration of compound present) measured in the serum of dogs. The compound shown at the bottom (highlighted in yellow) has the weakest inhibitory power (measured by IC50) but by far the best bioavailability (measured by cmax). This compound was selected for further development, leading to the drug indinavir (Crixivan).
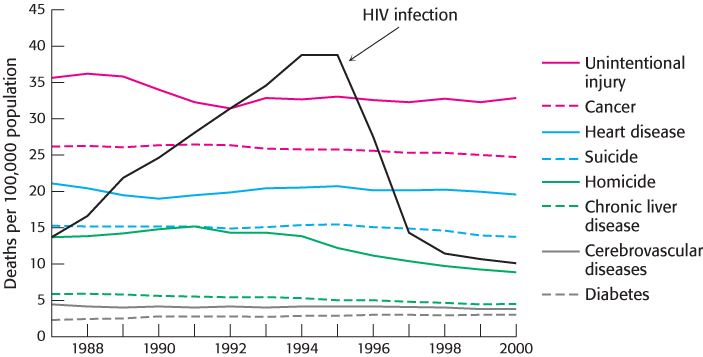
FIGURE 36.23The effect of anti-HIV drug development. Death rates from HIV infection (AIDS) reveal the tremendous effect of HIV protease inhibitors and their use in combination with inhibitors of HIV reverse transcriptase. The death rates in this graph are from the leading causes of death among persons 24 to 44 years old in the United States.
[Data from Centers for Disease Control.]
Aspirin targets the cyclooxygenase site in prostaglandin H2 synthase, as discussed earlier. Animal studies suggested that mammals contain not one but two distinct cyclooxygenase enzymes, both of which are targeted by aspirin. The more recently discovered enzyme, cyclooxygenase 2 (COX2), is expressed primarily as part of the inflammatory response, whereas cyclooxygenase 1 (COX1) is expressed more generally. These observations suggested that a cyclooxygenase inhibitor that was specific for COX2 might be able to reduce inflammation in conditions such as arthritis without producing the gastric and other side effects associated with aspirin.
The amino acid sequences of COX1 and COX2 were deduced from cDNA cloning studies. These sequences are more than 60% identical, clearly indicating that the enzymes have the same overall structure. Nevertheless, there are some differences in the residues around the aspirin-binding site. X-ray crystallography revealed that an extension of the binding pocket was present in COX2, but absent in COX1. This structural difference suggested a strategy for constructing COX2-specific inhibitors—namely, to synthesize compounds that had a protuberance that would fit into the pocket in the COX2 enzyme. Such compounds were designed and synthesized and then further refined to produce effective drugs familiar as Celebrex and Vioxx (Figure 36.24). Vioxx was subsequently withdrawn from the market because some people experienced adverse effects. Some of these effects may be due to the inhibition of COX2, the intended target. Thus, although the development of these drugs is a triumph for structure-based drug design, these outcomes highlight the fact that the inhibition of important enzymes can lead to complex physiological responses.
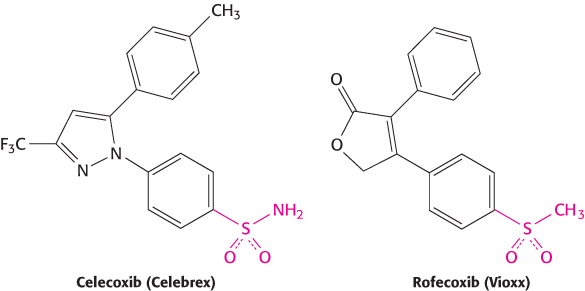
FIGURE 36.24COX2-specific inhibitors. These compounds have protuberances (shown in red) that fit into a pocket in the COX2 isozyme but sterically clash with the COX1 isozyme.