4.3 TERTIARY AND QUATERNARY STRUCTURES
Despite the use of only a few types of regular secondary structure, proteins exhibit a diverse spectrum of three-dimensional shapes, honed by evolution to perform their particular roles in the cell. And despite the great diversity of protein structure and function, general principles of protein shape apply. In overall shape, proteins are of two main types: the globular proteins, found in the aqueous environment of cells and in membranes, and the fibrous structural proteins, such as keratin and collagen. We concern ourselves here mainly with globular proteins. Globular proteins are roughly spherical, but contain sufficient irregularities to yield a surface area that is about twice that of a perfect sphere of equivalent volume. These nooks and crannies often form active sites or protein-protein interaction surfaces and are essential to protein function.
Knowledge of the tertiary and quaternary structures of a protein at atomic resolution offers a wealth of information and greatly helps our understanding of how the protein functions. Further, with a structure in hand, the biochemist can direct amino acid substitutions to defined architectural positions to test hypotheses about function. Atomic resolution of proteins that have medical relevance also enables the design of drugs aimed specifically at an active site.
Tertiary and Quaternary Structures Can Be Represented in Different Ways
The tertiary structure of a protein is defined by the three-dimensional orientation of all the different secondary structures and the turns and loops that connect them. The quaternary structure of a protein is defined by the connections between two or more polypeptide chains. A common quaternary structure is the association of two identical subunits. A protein consisting of two polypeptide subunits is called a dimer.
The tertiary and quaternary structures of the E. coli DNA polymerase β subunit, a dimer of two identical polypeptides that encircles the bacterial DNA, are shown in Figure 4-10. The β subunit is part of the replication apparatus that duplicates the genome, and it holds the apparatus to the DNA (we discuss DNA replication and the role of the β subunit in Chapter 11). The figure shows six different representations of the β dimer. Each representation emphasizes one or more of the many structural features of a complete protein, and no single diagram can represent them all. In most of the representations, two colors differentiate the two identical polypeptide chains that comprise the complete protein.
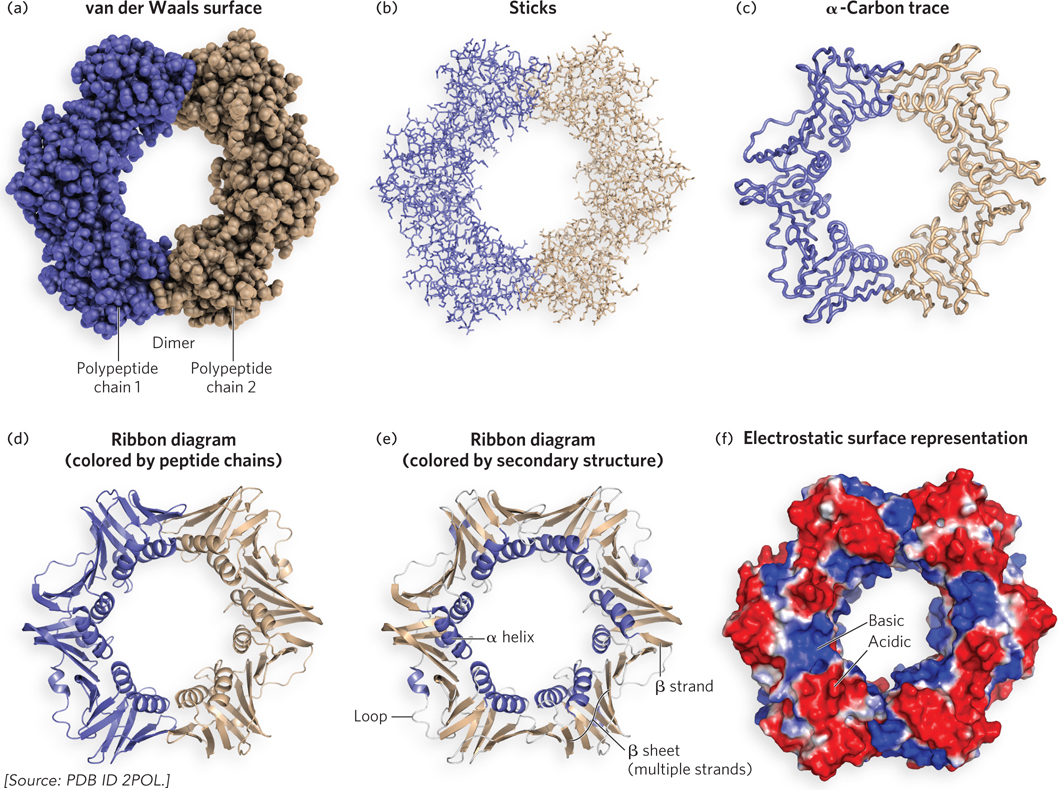
Figure 4-10: Different representations of tertiary and quaternary structure of the E. coli DNA polymerase β subunit. This protein dimer surrounds the E. coli DNA (not shown here).
Figure 4-10a shows the van der Waals radius for each atom, but because there are thousands of atoms in the structure, only the surface of the protein is visible. Figure 4-10b shows the atoms as sticks, allowing us to view the inside, but it is still somewhat bewildering to view the protein all at once. Typically, this representation is used to study the structure of just a small section of a protein. Figure 4-10c shows a thick-line trace of only the α carbons in the backbone, which simplifies the structure considerably and gives a view of the overall architecture. Figure 4-10d is a ribbon diagram, in which β sheets are shown as broad arrows pointing in the N- to C-terminal direction, β helices as coils, and loops and turns as narrow tubes, with the two polypeptides in different colors. The ribbon representation summarizes the secondary structural elements and overall architecture of the protein. Figure 4-10e is also a ribbon diagram, with the α helices and β sheets colored differently. Finally, Figure 4-10f is an electrostatic surface representation, showing the surface charges (red for acidic, blue for basic). The location of DNA-binding sites in proteins is often revealed by a basic patch in the electrostatic surface representation (note the blue area inside the β dimer).
Domains Are Independent Folding Units within the Protein
For a protein with 150 to 200 amino acid residues (Mr ∼20,000), the polypeptide chain usually folds into two folding units known as domains. The larger the protein, the more domains it usually contains. The secondary structures that comprise a domain are typically adjacent to one another in the primary sequence, although this is not always the case. A protein with two or more domains may perform a single function in the cell, but sometimes, different domains within one protein have different functions. Although domains are independent folding units that can form a folded structure when separate from the full-length protein, various domains in the same protein often interact. The boundaries between domains are usually assessed by examining the protein’s structure, but they can also be defined by limited proteolysis (a technique that cleaves the polypeptide backbone), which sometimes separates a protein into its domains.
Domains, then, can have independent functions. For example, the zinc finger domain is often used to bind DNA. (See this chapter’s Moment of Discovery and Figure 4-20a; also see Chapter 19 for more on zinc-binding domains.) An individual domain can also catalyze a reaction, such as a nuclease activity. There are also proteins that require several domains that come together to perform a single function. Structures of proteins containing one, two, three, and four domains are shown in Figure 4-11. Myoglobin (Mr 16,700) is a small, oxygen-binding protein that folds into a single domain. An example of a two-domain protein is γ crystallin (Mr 21,500), a component in the lens of the eye. DnaA (Mr 52,000), a bacterial protein involved in the initiation of DNA synthesis, contains three domains. A proteolytic fragment of E. coli DNA polymerase I (Mr 68,000), an enzyme that synthesizes new DNA strands, consists of four domains with two separate enzymatic activities. One domain comprises an exonuclease, which proofreads the product of the DNA polymerase and removes any mistakes. The other three domains cooperate to form the DNA polymerase activity. (DNA polymerase structure and function are described in Chapter 11.)
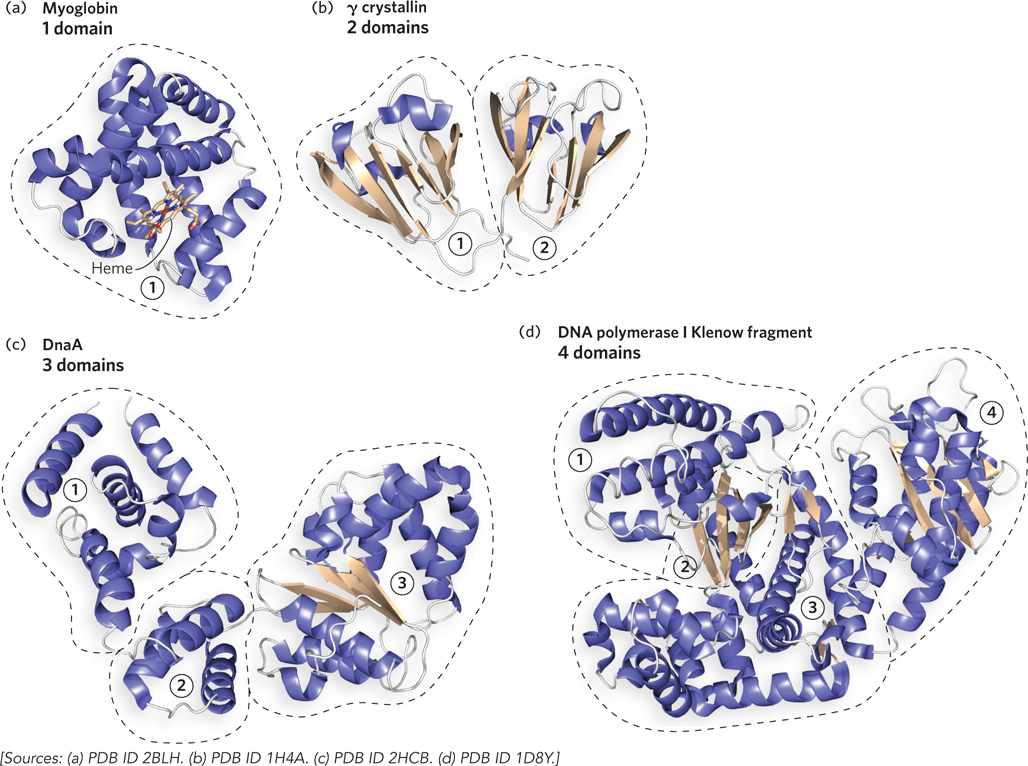
Figure 4-11: Domains. Proteins fold into one or more domains, depending on their size. (a) One domain: sperm whale myoglobin. (b) Two domains: human γ crystallin. (c) Three domains: Aquifex aeolicus DnaA. (d) Four domains: E. coli Pol I Klenow fragment; the DNA polymerase activity is contained in the domains numbered 1, 2, and 3, and the exonuclease activity is in the fourth domain.
Supersecondary Structural Elements Are Building Blocks of Domains
Particularly stable and common arrangements of multiple secondary structural elements are called supersecondary structures, also referred to as structural motifs or folds. Supersecondary structures are linked together to form sections of domains, or even whole domains. Some supersecondary structures are formed only of β sheets, some only of α helices, and others have a combination of α helices and β sheets.
Structural Motifs Containing a β Sheet The smallest motif containing a β sheet is the β hairpin, in which two antiparallel β strands are connected, often by a β or γ turn (Figure 4-12a). In proteins that contain this motif, the β hairpin can be found alone or as a repeated structure that forms a larger, antiparallel β sheet. Whether they are parallel or antiparallel, β sheets tend to follow a right-handed twist (Figure 4-12b). When the strands of a β sheet contain hydrophobic R groups at every second residue, the groups lie on the same side of the sheet, thus facilitating layer formation in the folded state. For example, a β sheet with a hydrophobic surface may pack against the hydrophobic side of another sheet. A β sheet of eight or more strands, and with one surface that is hydrophobic, can form a cylinder in which the first β strand hydrogen-bonds with the last β strand. This structure, referred to as a β barrel, sequesters the hydrophobic side chains inside the cylinder (Figure 4-12c). In proteins with a β barrel, the barrel is a single supersecondary structure that also forms a complete domain. The simplified diagram below each supersecondary structure in Figure 4-12 shows the chain-folding pattern of the structure in two dimensions and is known as a chain topology diagram.
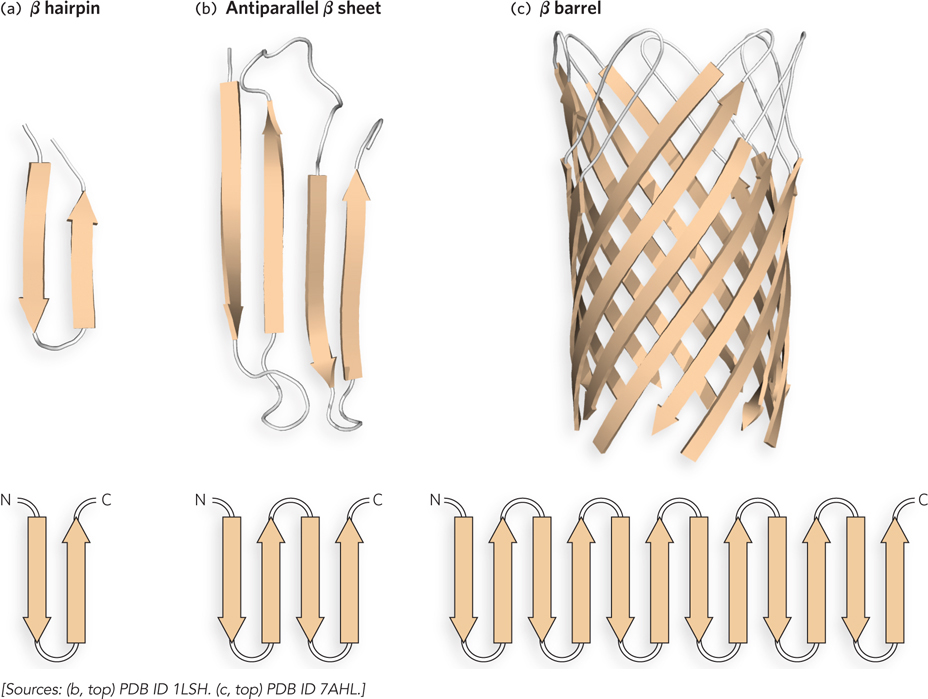
Figure 4-12: Supersecondary structures of antiparallel β sheets. (a) The β hairpin motif is an antiparallel β sheet composed of two β strands adjacent in the primary structure; the strands are often connected by β or γ turns. (b) The β strands of antiparallel (shown here) and parallel β sheets tend to have a right-handed twist. (c) The β barrel motif is formed by the twist in a β sheet with a connection between the first and last strands. This example is a single domain of hemolysin (a pore-forming toxin that kills a cell by creating a hole in its membrane) from the bacterium Staphylococcus aureus. The diagram below each structure shows the folding topology of the polypeptide chain. The β strands in a β barrel can have several different chain topologies; shown in (c) is an “up-and-down barrel,” reflecting the chain topology. In a simple β barrel, the strands would be connected as shown in the topology diagram (bottom), but the bottom strands of the particular β barrel shown here (top) are not connected, because the barrel is associated with additional domains (not shown).
The Greek key motif, named for a design on Greek pottery, is another common β motif, consisting of four antiparallel β strands (Figure 4-13). One example is found in the OB-fold domain, which mediates DNA binding in many different types of proteins. We will encounter the OB fold again in Chapter 5, where the functions of many DNA-binding proteins are discussed.

Figure 4-13: The Greek key motif. (a) A portion of the subunit of E. coli DNA polymerase III adopts the Greek key conformation. (b) A chain topology diagram of the Greek key motif. (c) An example of the design found on Greek pottery from which this motif derives its name.
Motifs with α Helix and α Helix/β Sheet Structures In parallel β sheets, the connection between adjacent strands requires a much longer linker than in antiparallel β sheets. A basic unit of parallel β sheets is the β-α-β motif, which consists of two parallel β strands connected by an α helix. The β-α-β motifs can be stitched together by the α helix linker in two different ways. In one, the α helix linker connects two β strands that are adjacent and hydrogen-bonded to each other (Figure 4-14a). This linkage forms a very common domain architecture called an α/β barrel, consisting of eight β strands surrounded on the outside by eight α helices (Figure 4-14b). The active site of an α/β barrel is almost always found on the loops at one end of the barrel.
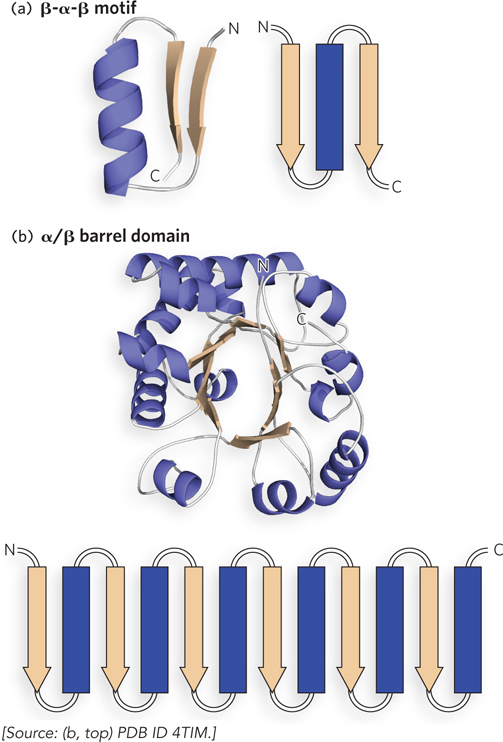
Figure 4-14: The β-α-β motif. (a) The β-α-β motif contains two parallel β strands connected by an α helix. (b) Four β-α-β motifs, interconnected by α helices, underlie the α/β barrel structure. This α/β barrel is from triosephosphate isomerase of Trypanosoma brucei. The topology diagram in (b) shows the general arrangement of β strands and α helices; the actual structure of this α/β barrel domain contains eight β strands connected by α helices.
The second way that the β strands in a β-α-β motif are joined by an α helix is shown in Figure 4-15. This arrangement does not allow β strands in adjacent β-α-β motifs to hydrogen-bond and therefore prevents circularization of the β-α-β motifs. The result is a domain with a central parallel β sheet that contains α helices on both sides of the sheet. This architecture is commonly observed in ATP- or GTP-binding proteins and is sometimes referred to as a Rossmann fold. The active site in these proteins is usually located on the loops at the junction formed by β strands that are not directly connected by an α helix (see the How We Know section at the end of this chapter).
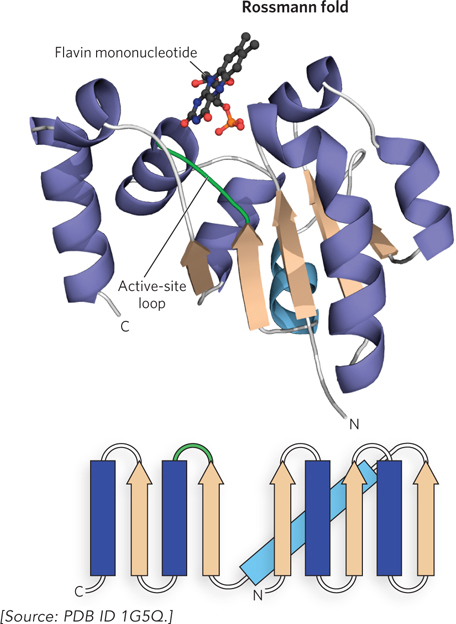
Figure 4-15: The nucleotide-binding Rossmann fold. The Rossmann fold, a common nucleotide-binding motif, is minimally composed of a β-α-β-α-β motif. This example is from a section of EpiD, a decarboxylase enzyme from the bacterium Staphylococcus epidermis. The β-α-β motifs are connected as shown in the chain topology diagram (bottom). In EpiD, the active-site loop binds a flavin mononucleotide.
Several motifs have only α helices. One supersecondary structure that uses two α helices is the helix-turn-helix motif, sometimes found in proteins that bind specific DNA sequences (Figure 4-16a). It is commonly found in bacterial transcription factors (proteins that regulate gene expression), as well as in some eukaryotic transcription factors. Another common α helix supersecondary structure is the four-helix bundle (Figure 4-16b). This structure consists of four α helices; the way they interact depends on the protein. Sometimes the bundle is formed by antiparallel helices, and other times by parallel helices or a mixture of the two. Some four-helix bundles are even formed by dimerization of two different subunits, each of which contributes two α helices to the motif. In the coiled-coil motif, two α helices pack against each other at an angle of 18° and gently twist around one another in a left-handed supercoil (Figure 4-16c). The two α helices interact through hydrophobic contacts along the sides of each helix that form the coiled-coil interface.
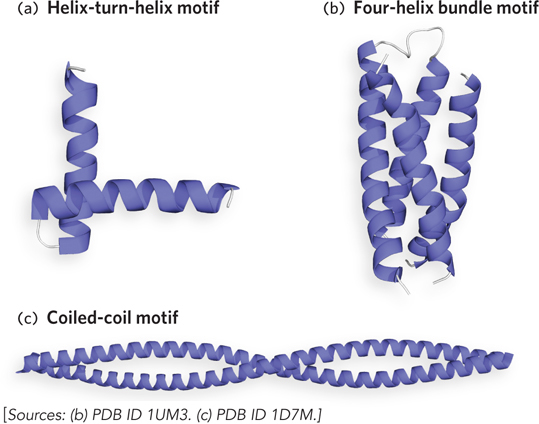
Figure 4-16: Supersecondary structures of α helices. (a) The helix-turn-helix motif is common in DNA-binding proteins. (b) The four-helix bundle is formed by the interaction of four α helices. (c) The coiled-coil motif consists of two α helices twisted in a left-handed supercoil.
The interaction between α helices in a protein occurs through hydrophobic surfaces that face one another. A useful way to visualize the alignment of residues along the edges of an α helix is the helical wheel, a two-dimensional representation of the residues in the α helix (Figure 4-17a). The α helix has a nonintegral number of residues (3.6) per turn, but a nearly integral number of residues (7.2) in two turns. For this reason, the helical wheel representation consists of seven positions along two turns of α helix, designated by letters a through g.
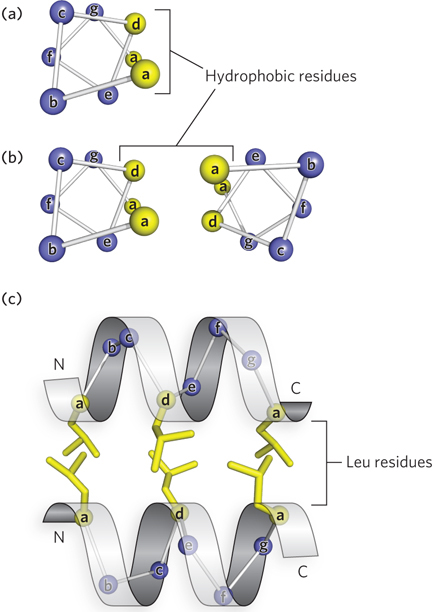
Figure 4-17: The helical wheel and heptad repeats. (a) The seven residues that comprise two turns of the α helix can be represented by a helical wheel and are labeled a through g. (b) Residues in positions a and d lie on the same side of the helix; hydrophobic residues in these positions create an amphipathic helix and form the interface with a second helix to form a coiled-coil. (c) The packing of a and d residues of two α helices in a coiled-coil. Here, the hydrophobic residues are leucines.
Residues that are spaced every two turns, or every seven residues, lie on approximately the same side of the helix. When two α helices interact with each other by forming a hydrophobic interface, they often contain a repeating pattern of a hydrophobic residue every seventh amino acid. This seven-residue spacing pattern of hydrophobic residues in an α helix is sometimes referred to as a heptad repeat, or a leucine repeat, because the residue occupying the d position in a heptad repeat such as this is often leucine. Coiled-coils usually contain two heptad repeats, one set within the other, with a-to-a and d-to-d spacing that allows contact with another helix with a similar arrangement of hydrophobic residues, as illustrated by the α-helical wheel diagrams in Figure 4-17b. This four-and-three hydrophobic repeat pattern is a hallmark of coiled-coils and forms a helix that is hydrophobic on one side and hydrophilic on the rest of the surface, which is referred to as an amphipathic helix (Figure 4-17c).
Some proteins, such as keratin, are long, extended, fibrous proteins in which the main structural element is a very long coiled-coil. However, there are many examples of globular proteins with short coiled-coils that mediate dimer formation. For example, the common leucine zipper motif in some eukaryotic transcription factors consists of four to five heptad repeats of leucine. These globular proteins, each containing a leucine zipper motif, can dimerize.
Quaternary Structures Range from Simple to Complex
A protein composed of multiple polypeptide chains is referred to as an oligomer, or multimer, and the individual polypeptide chains are referred to as subunits, or protomers. An oligomer with identical subunits is referred to as a homooligomer, while an oligomer with nonidentical subunits is a heterooligomer. The quaternary structures of three proteins are shown in Figure 4-18. An example of a homodimer (i.e., a protein with two identical subunits) is E. coli cAMP receptor protein (CRP), also called catabolite gene activation protein (CAP) (Mr 22,000). Each CRP subunit has two domains; one domain binds DNA and the other binds the cyclic nucleotide cAMP. In the homodimer, the two CRP protomers interact through a coiled-coil, and the two DNA-binding domains are oriented adjacent to each other (Figure 4-18a). An example of a homotrimer is eukaryotic PCNA, in which the three subunits (each subunit Mr 29,000) are joined by an intermolecular β sheet and by helix-helix packing (Figure 4-18b). PCNA functions like the E. coli β subunit, anchoring the replication apparatus to DNA. Hemoglobin is a well-studied example of a heterooligomer (tetramer Mr 64,500). Hemoglobin contains two a protomers and two β protomers (Figure 4-18c).
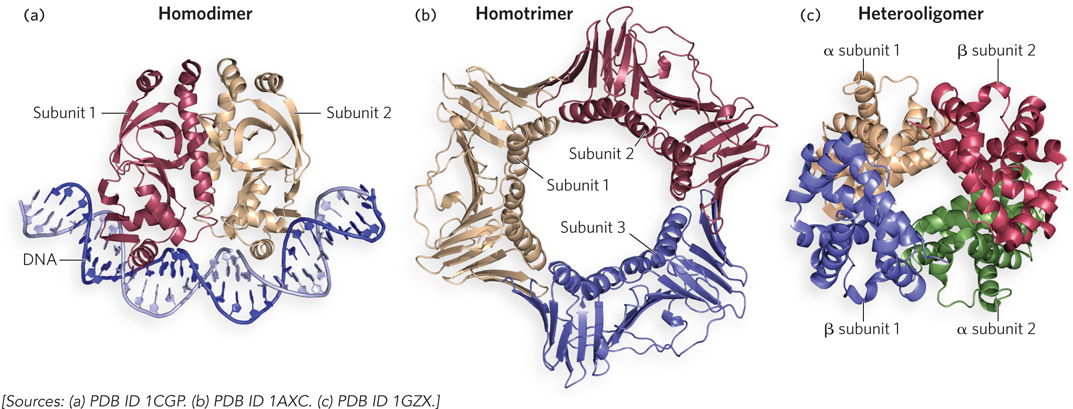
Figure 4-18: The quaternary structure of three proteins. Protomers (subunits) are represented in different colors. (a) Homodimeric E. coli CRP protein, as a complex with DNA. (b) Human PCNA, a homotrimer. (c) Human hemoglobin, a heterooligomer of two α chains and two β chains.
Some oligomers are made up of numerous subunits. One example is the ribosome, a large, multiprotein oligomer that contains many nonidentical subunits, each often present as a single copy, as well as functional RNAs—all of which, together, form a molecular machine that translates a nucleic acid sequence into a protein sequence. The GroEL chaperonin is another example of a large, machinelike oligomer, which we discuss in Section 4.4.
Why do cells assemble such large oligomers from multiple subunits, rather than producing a large protein as a single, multidomain polypeptide chain? There are several reasons. For one, the folding of the many different domains in a single, very large polypeptide chain may be problematic. In addition, if one domain did not fold properly, the entire protein would lack function, so the investment of energy in making it would be wasted. A multisubunit composition avoids these problems. If a protein subunit misfolds, it will not be included in the oligomer, but at least only the investment of cellular resources to make this one domain will be wasted. In fact, a similar argument can be made even if the entire machinery is folded correctly. If one subunit subsequently denatures (becomes unfolded) or becomes inactive for any reason, it can be replaced by another subunit. The accuracy of translation is also an issue for very large proteins. During protein synthesis, approximately one mistake occurs in every 100,000 peptide-bond joining events. Therefore, a large protein of Mr exceeding 106 may accumulate mistakes, perhaps becoming inactive; smaller, individual subunits that do not fold properly can simply be discarded. Finally, the architecture of many large oligomers includes multiple copies of some subunits. More DNA would be required to encode a single large protein than is needed to encode multiple copies of individual subunits.
Intrinsically Unstructured Proteins Have Versatile Binding Properties
In the past 10 years, a very unconventional and new class of protein has been discovered that functions without having a defined structure. These intrinsically unstructured proteins appear to carry out some of the most important duties of the cell. Intrinsically unstructured proteins lack any detectable tertiary structure at all and normally would be regarded as inactive or denatured protein. Yet, because of their unstructured state, they are capable of binding to several different types of sites on different partners, and thus the same protein can be reused in multiple pathways. This diversity in binding is believed to play important roles in cell signaling, placing this class of protein at the center of protein interaction networks, where it can act as a hub for information transmission. Disease datasets indicate that intrinsically unstructured proteins may underlie pathological states with altered regulation of signaling, transcriptional control, cell division, and translation. Although an unstructured protein may acquire some structure on binding another protein, this is not always so. It is also thought that many normally structured proteins have a section of unstructured polypeptide, of 50 residues or more, that may serve the same purpose—to bind other proteins. One might think that this class of protein would be relegated to a very small niche of proteins encoded by a genome. But this is not the case. In fact, the prevalence of proteins, or sections within proteins, that fall into this category increases with the evolutionary sophistication of the organism. It has been proposed that up to one-third of proteins in the more complex eukaryotes either are unstructured or contain an intrinsically unstructured region. Indeed, the ability of this type of protein to bind many partners and act as an information network hub extends the complexity that can be extracted from the limited amount of DNA that can be packaged into a cell.
Protein Structures Help Explain Protein Evolution
Proteins with a similar primary sequence and similar function usually share a common evolutionary heritage and are said to be in the same protein family. For example, the globin family consists of many different proteins with structural and sequence similarity to myoglobin (e.g., myoglobin and the α and β subunits of hemoglobin have the same folding pattern). In many cases, however, even though the primary sequence does not show an evolutionary relationship, the protein structures are similar, indicating that they are related through a common ancestor. This is because the three-dimensional structure of a protein is more highly conserved than the primary sequence.
Protein families with little sequence similarity but with the same supersecondary structural motif and functional similarities are referred to as superfamilies. An evolutionary relationship among the families of a superfamily is considered probable, even though time and functional distinctions—resulting from different adaptive pressures—may have erased many telltale sequence relationships. A protein family may be widespread in all three domains of life—the Bacteria, Archaea, and Eukarya—suggesting a very ancient origin. For example, members of the α/β hydrolase superfamily contain eight β strands connected by α helices with three catalytic residues occurring in a particular order. This supersecondary structure is found in various proteases, esterases, lipases, and other types of proteins. Other families may be present in only a small group of organisms, indicating that the protein structure arose more recently. An example of this is the eukaryotic Ras superfamily of small GTPases, members of which contain a conserved “G domain” and are involved in cell proliferation, vesicle transport, and cell morphology. Tracing the natural history of structural motifs, using systematic structural classification databases, provides a powerful complement to sequence analysis (Highlight 4-2).
HIGHLIGHT 4-2 A CLOSER LOOK: Protein Structure Databases
Each protein structure contains thousands of atoms arranged in three-dimensional space. When a scientist determines a protein structure, the x, y, and z coordinates for each atom are stored in a database called the protein Data Bank (PDB) and each is assigned a PDB identification number (ID). To view the atomic structure of a protein on a computer screen, the PDB ID file of atomic coordinates is imported into a computer program that displays the atomic model. There are many types of computer programs written just for this purpose, including PyMol, which is easily used on desktop and laptop computers.
Proteins can be classified according to their secondary structural elements, supersecondary structural motifs, and sequence homologies, and there are several databases that classify protein structures. One of the oldest is the Structural Classification of Proteins (SCOP) database. The highest level of classification in the SCOP database places all proteins into one of four classes: all α helix, all β sheet, α/β (where α and β segments are interspersed), and α + β (where α and β segments are somewhat segregated). For instance, myoglobin has α helices but no β sheet and therefore is placed in the α class. Likewise, proteins consisting only of a β barrel are in the β class. DnaA and Pol I have α helices mixed in with β sheets and thus are in the α/β class. In the circular β and PCNA clamps, the β sheet and α helices are somewhat segregated, placing these proteins in the α + β class.
Each class contains tens to hundreds of distinct substructure folding arrangements. Some substructures are very common, and others are found in only one protein. The number of unique folding motifs in proteins is far lower than the number of proteins—perhaps fewer than 1,000 different folds. As new protein structures are elucidated, the proportion of those containing a new structural motif has been declining. Below the levels of class and fold (or motif), which are purely structural, categorization in the SCOP database is based on evolutionary relationships.
The SCOP database is curated manually, with the objective of placing proteins in the correct evolutionary framework on the basis of conserved structural features. Two similar enterprises, the CATH (Class, Architecture, Topology, and Homologous superfamily) and FSSP (Fold Classification Based on Structure-Structure alignment of Proteins) databases, make use of more automated methods and can provide additional information.
SECTION 4.3 SUMMARY
The tertiary structure of a protein is its three-dimensional structure, consisting of all of its secondary structural elements and loops.
A protein domain is an independent folding unit within a protein and typically consists of up to 150 residues.
Supersecondary structural elements, also called structural motifs, are arrangements of multiple secondary structural elements commonly found in proteins. Supersecondary structures are the building blocks associated with particular functions.
The quaternary structure of a protein includes all the connections between two or more polypeptides and can range from a simple homodimer to a large, multiprotein assembly such as a ribosome.
Some proteins have no apparent tertiary structure, yet are known to exist in cells and function to organize other proteins.
The three-dimensional structure of proteins that evolved from a common ancestor is often more conserved than the primary sequence, making protein structures very useful in determining evolutionary heritage.