DNA Molecules Have Distinctive Base Compositions
In the 1940s, Erwin Chargaff and his colleagues made an important discovery that provided clues to the structure of DNA. Using DNA samples isolated from many different organisms, they observed that the four bases of DNA occur in different ratios that are characteristic of each species. They also observed that for specific pairs of bases, the amounts of each base are closely related. Their data showed that:
The base composition of DNA generally varies from one species to another.
DNA specimens isolated from different tissues of the same species have the same base composition.
The base composition of DNA in a given species does not change with an organism’s age, nutritional state, or environment.
In all cellular DNAs, regardless of the species, the number of adenosine residues equals the number of thymidine residues (i.e., A = T), and the number of guanosine residues equals the number of cytidine residues (G = C). From these relationships it follows that the sum of the purine residues equals the sum of the pyrimidine residues: A + G = T + C.
Referred to as Chargaff’s rules, these quantitative relationships were confirmed by many subsequent researchers. Not only were these findings a key to establishing the three-dimensional structure of DNA, but they also yielded clues about how genetic information is encoded in DNA and passed along from one generation to the next.
DNA Is Usually a Right-Handed Double Helix
Chargaff’s discoveries in the 1940s imposed important constraints on possible models for the structure of DNA. At the same time, Rosalind Franklin and Maurice Wilkins were using the powerful method of x-ray diffraction to analyze DNA fibers (see the How We Know section at the end of this chapter). Franklin’s data showed that DNA produces a characteristic x-ray diffraction pattern. From this pattern, Watson and Crick deduced that DNA molecules are helical, with two periodicities along their long axis: a primary one of 3.4 Å and a secondary one of 34 Å. The challenge then was to formulate a three-dimensional model of the DNA molecule that could account not only for the x-ray diffraction data but also for the specific A = T and G = C base equivalences discovered by Chargaff.
The x-ray diffraction data obtained by Franklin and Wilkins enabled Watson and Crick to formulate a model in which DNA is composed of two polynucleotide strands entwined in the form of a right-handed double helix (Figure 6-14). Alternating 2′-deoxy-d-ribose and phosphate units make up the backbone of each strand, from which the bases project inward, toward the center of the helix. The bases are thus positioned to form hydrogen-bonding interactions with each other according to the preferred pairings of A with T and G with C. The twist of the helix forms two unequal surfaces, which are called the major groove and the minor groove. DNA strands always have a defined directionality, or polarity, due to the asymmetric shape and chemical linkage of the component nucleotides. In the double helix, the two strands have opposite directionality, and the helix is said to be antiparallel. In chemical terms, this means that one strand runs in the 5′→3′ direction and the other runs in the 3′→5′ direction. The antiparallel orientation of the DNA strands is more energetically favorable than the parallel orientation, due to the geometry of the component bases. Furthermore, the DNA double helix almost always twists in a right-handed direction (see Figure 4-6b for an explanation of helix handedness). Rarely, left-handed helices are observed in DNA, with the twist to the left. By convention, DNA helices are assumed to be right-handed unless otherwise specified.
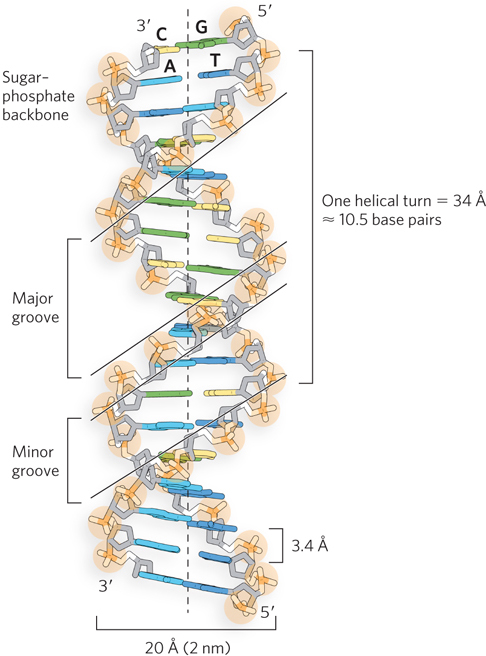
Figure 6-14: The double-helical structure of DNA. The original model proposed by Watson and Crick had 10 base pairs and a length of 34 Å per turn of the helix. Later measurements of DNA in solution (as opposed to in a crystal or fiber) showed 10.5 base pairs per helical turn. The major and minor grooves, where most interactions with proteins or other nucleic acids occur, are shown.
Watson and Crick’s double-helical model of DNA makes chemical sense, accounting for the known properties of the component nucleotides. The hydrophilic backbones of alternating sugar and phosphate groups are on the outside of the helix, facing the surrounding water. The pentose ring of each deoxyribose is in the C-2′ endo conformation (see Figure 6-3), and this sugar pucker defines the distance between adjacent phosphate groups in the DNA backbone. The purine and pyrimidine bases of both strands are stacked inside the double helix, with their hydrophobic and nearly planar ring structures very close together and more or less perpendicular to the long axis. Each nucleotide base of one strand is paired in the same plane with a base of the other strand. Watson and Crick found that the hydrogen-bonded base pairs, A with T and G with C, are those that agreed best with the x-ray diffraction data, providing a rationale for Chargaff’s rule that in any DNA, A = T and G = C. It is important to note that three hydrogen bonds can form between G and C, symbolized G≡C, but only two can form between A and T, symbolized A=T. By always pairing a purine (A or G) with a pyrimidine (T or C), consistent spacing is maintained between the two antiparallel DNA backbones, giving a regular, uniform shape to the double helix. This has significant consequences for the stability of any double-stranded DNA sequence, as we will see in Section 6.4.

Rosalind Franklin, 1920–1958

Maurice Wilkins, 1916–2004
The double-helical structure of DNA also explains the periodicities observed in the x-ray diffraction patterns of DNA fibers. The vertically stacked bases inside the double helix are 3.4 Å apart; the secondary repeat distance of about 34 Å is accounted for by the presence of 10 base pairs in each complete turn of the double helix. In aqueous solution the structure differs slightly from that in fibers, having 10.5 base pairs per helical turn.
The stability of the DNA double helix arises primarily from the hydrophobic base-stacking interactions, which are largely nonspecific with respect to sequence. The configuration of planar purine–pyrimidine base pairs at the center of the helix allows their flat surfaces to stack on top of each other (see Figure 6-10), through dipole-dipole interactions and pi orbital stacking (discussed in Chapter 3). This energetically favorable situation stabilizes the double helix relative to single-stranded DNA by minimizing contact of the hydrophobic purines and pyrimidines with water. Furthermore, extensive networks of weak bonds in double-stranded DNA, such as van der Waals interactions and hydrogen bonds, are arranged so that for most of these bonds, they cannot break without simultaneously breaking many others. Consequently, DNA double helices that are 10 or more base pairs long are quite stable at room temperature. As mentioned in Section 6.1, DNA can persist in fossil samples over long periods of time, making possible the sequencing of DNA samples from long-extinct species—including Neanderthal hominids and woolly mammoths.
The most significant property of the double helix as an information carrier is the hydrogen bonding between the bases. Because adenine is always hydrogen-bonded to thymine, and guanine is always hydrogen-bonded to cytosine, exact copies of encoded information can be replicated. This specific base pairing gives the two helical strands a complementary relationship in which the base sequence of one strand defines the sequence of its partner. For example, the sequence 5′-GTAACGC-3′ on one strand specifies the complementary sequence 5′-GCGTTAC-3′ on the other strand.
Thus, the discovery of the DNA double helix immediately suggested a mechanism for the transmission of genetic information. As Watson and Crick proposed, this structure could logically be reproduced by separating the two strands and synthesizing a complementary strand for each. Because nucleotides in each new strand are joined in a sequence specified by the base-pairing rules stated earlier, each preexisting strand functions as a template to guide the synthesis of a complementary strand (Figure 6-15). These expectations were experimentally confirmed, inaugurating a revolution in our understanding of biological inheritance. (DNA replication is discussed in detail in Chapter 11.)
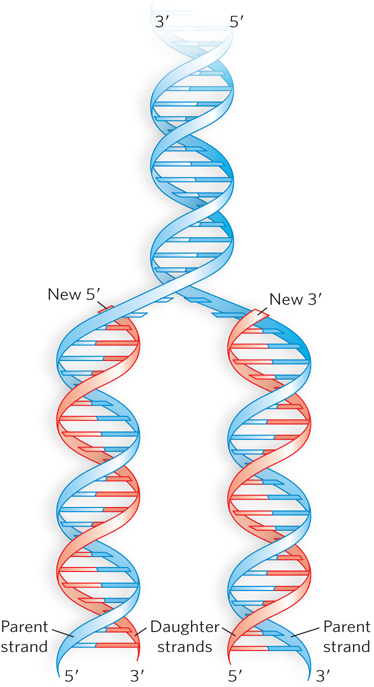
Figure 6-15: The mechanism for DNA replication. The newly synthesized complementary strands (“daughter” strands) are shown in red.
DNA Adopts Different Helical Forms
Nucleic acids are inherently flexible molecules. Numerous bonds in the sugar–phosphate backbone can rotate, and thermal fluctuation can lead to bending, stretching, and localized unpairing of the two strands. As a result, cellular DNA contains significant deviations from the Watson-Crick DNA structure, some or all of which may play important roles in DNA metabolism. Generally, such structural variations do not affect the key properties of strand complementarity: antiparallel strands and the requirement for A=T and G≡C base pairs.
Variation in the three-dimensional structure of DNA reflects three things: the different possible conformations of the deoxyribose (see Figure 6-3), rotation about the contiguous bonds that make up the sugar–phosphate backbone (Figure 6-16a), and free rotation about the glycosidic bond. Because of steric constraints, purines in purine nucleotides are restricted to two stable conformations with respect to deoxyribose, called syn and anti (Figure 6-16b). Pyrimidines are generally restricted to the anti conformation, because of steric interference between the sugar and the carbonyl oxygen at C-2 of the pyrimidine.
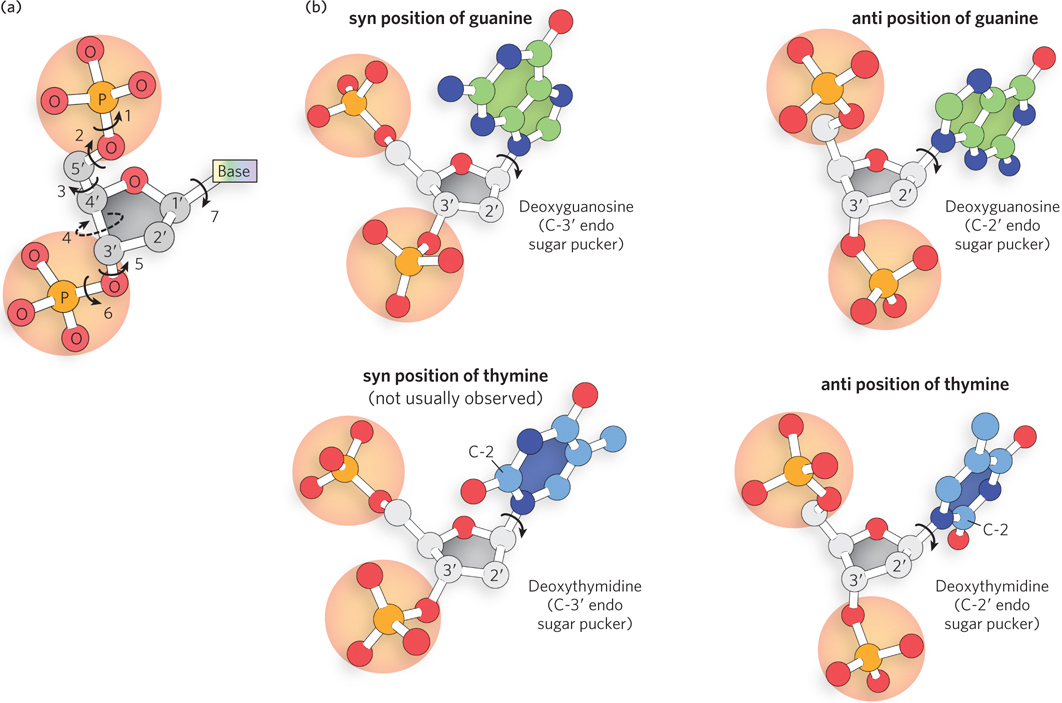
Figure 6-16: Factors contributing to structural variation in DNA. (a) DNA nucleotide conformation is affected by rotation about seven different bonds. Six of the bonds rotate freely; rotation about bond 4 is constrained by the sugar ring, giving rise to the sugar pucker. (b) The syn and anti positions of deoxyguanosine, and the syn and anti positions of deoxythymidine. Note that pyrimidines are restricted to the anti position; the carbonyl at C-2 causes steric clash in the syn conformation.
The Watson-Crick structure is known as B-form DNA, or B-DNA. As the most stable structure for a random-sequence DNA molecule under physiological conditions, B-DNA is the standard point of reference in any study of the properties of DNA. Structures of short B-DNA duplexes have been studied in depth, revealing many details about the double helix (see the How We Know section at the end of this chapter). Two structural variants well characterized by x-ray crystallography are A-form DNA (A-DNA) and Z-form DNA (Z-DNA). These three DNA conformations are shown in Figure 6-17, with a summary of their properties.
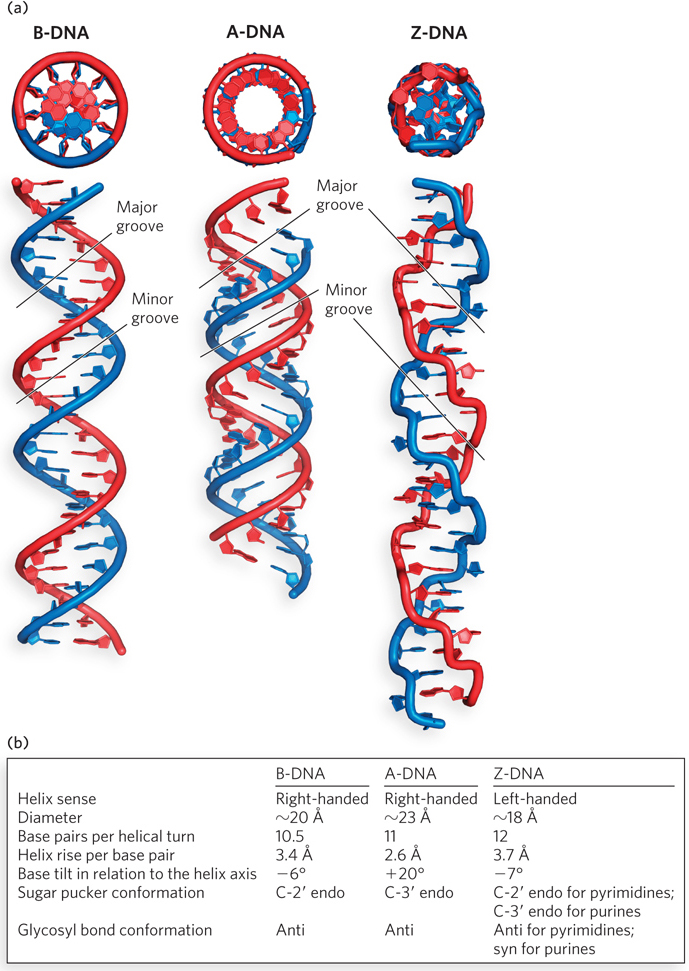
Figure 6-17: A comparison of the B, A, and Z forms of DNA. (a) In each case, the sugar–phosphate backbones wind around the exterior of the helix (red and blue), with the bases pointing inward. The same 25 base pair DNA sequence is shown in all three forms. (As described later in the chapter, double-stranded RNA typically assumes the A-form geometry shown here.) Differences in helical diameter can be seen in the end-on views (top); differences in helical rise and groove shape are apparent in the side views (bottom). B-DNA, the most common form in cells, has a wide major groove and a narrow minor groove. A-form helices, common for RNA and certain DNA structures, are more compact than B-DNA. The major groove is deeper and the minor groove is shallower than in B-DNA. Z-DNA, which forms only under high salt conditions or with C≡G-rich DNA sequences, is left-handed, and its backbone has a zigzag pattern. It is less compact than B-DNA, with a very shallow major groove and a narrow and deep minor groove. (b) The table summarizes some properties of the three forms of DNA.
A-DNA is favored in many solutions that are relatively devoid of water. In this case, the DNA is still arranged in a right-handed double helix, but the helix is wider (23 Å in diameter, compared with 20 Å for B-DNA) and the number of base pairs per helical turn is 11, rather than 10.5 as in B-DNA. Whereas base pairs in B-DNA tilt slightly in a negative direction—that is, below the plane—with respect to a plane perpendicular to the helical axis, the base pairs in A-DNA are tilted above the plane by about +20°. In addition, the distance between adjacent phosphates in the polynucleotide chain, a direct consequence of the sugar pucker, decreases from 7 Å in B-form helices to 5.9 Å in A-form helices (Figure 6-18). These structural changes deepen the major groove while making the minor groove shallower. The reagents used to promote crystallization of DNA tend to dehydrate it, and thus most short DNA molecules tend to crystallize in the A form.
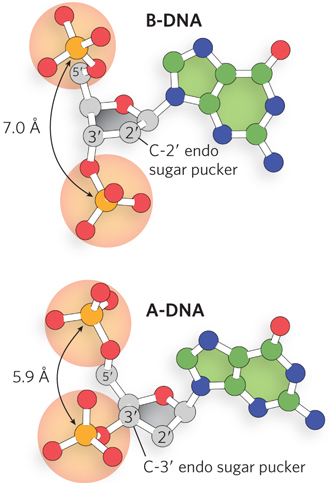
Figure 6-18: The effect of sugar pucker on the distance between phosphates in nucleic acids. The configuration of the pentose ring, known as the sugar pucker, is different in the backbones of B-DNA and A-DNA. As a result, the distance between the phosphate groups attached to the 5′ and 3′ positions of each nucleotide differs in the two forms, giving rise to distinct helical geometries.
Z-DNA is a more radical departure from B-DNA; the most obvious distinction is the left-handed helical rotation. There are 12 base pairs per helical turn, and the structure appears more slender and elongated. The DNA backbone takes on a zigzag appearance (hence the Z designation). Certain nucleotide sequences fold into left-handed Z helices much more readily than do others. Prominent examples are sequences in which pyrimidines alternate with purines, especially alternating C and G residues or 5-methyl-C and G residues (methylated bases are discussed in Section 6.4). To form the left-handed helix in Z-DNA, the purine residues flip to the syn conformation, alternating with pyrimidines in the anti conformation. The major groove is barely apparent in Z-DNA, and the minor groove is narrow and deep.
Whether A-DNA occurs in cells is uncertain, but there is evidence for some short stretches of Z-DNA in the chromosomes of both bacteria and eukaryotes. The evidence comes in part from experimentally prepared antibodies against short Z-form DNA segments, which can selectively bind to sequences in chromosomal DNA. Some evidence suggests that these potential Z-tracts correspond to actively transcribed regions of the genome, but it is also possible that antibody detection in these experiments is misleading due to capture of transient DNA structures or even spurious binding reactions. Thus, the function of these potential Z-DNA tracts remains unclear.
Certain DNA Sequences Adopt Unusual Structures
Other sequence-specific DNA structures have been detected, within larger chromosomes, that may affect the function and metabolism of the DNA segments in their immediate vicinity. For example, certain repetitive sequences can bend the DNA helix in a distinct way. The stability and geometry of base-pair stacking influences the preferred direction of DNA bending. This was first observed in repetitive-sequence DNA isolated from trypanosomes, the protozoa that cause African sleeping sickness. Typical sequences that cause pronounced bending contain stretches of four to six A and T residues separated by C- and G-rich segments. The A- or T-tracts, each corresponding to a half-turn of the double helix, are spaced such that the geometry of the repetitive base pairs tends to curve the DNA helix in one direction (Figure 6-19). This DNA bending helps certain proteins—such as transcription factors, which promote the synthesis of mRNAs—bind to their target DNA binding sites.
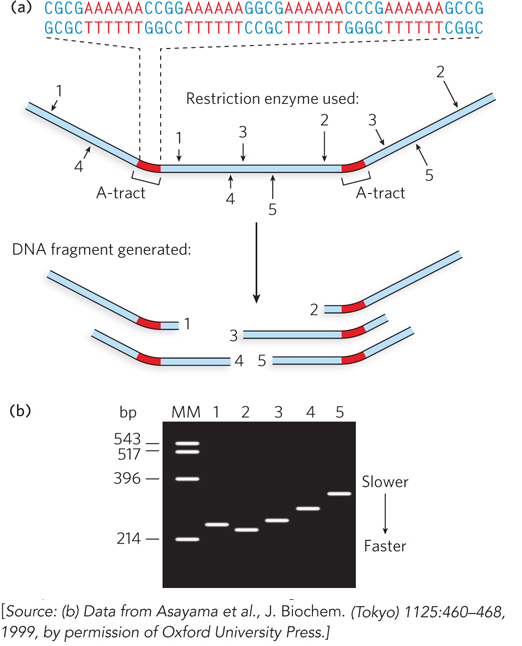
Figure 6-19: DNA bending at A-tracts. Bending in DNA segments containing A-tracts of six adenosines in a row can be detected by using enzymes to cut the DNA at specific sites. (a) The DNA sequence used in this experiment contains two A-tracts. Arrows point to sites that can be cleaved by various restriction enzymes (numbered 1 to 5) to generate DNA fragments of equal length that contain an A-tract at various positions in the fragment. (b) DNA fragments are analyzed by gel electrophoresis. Even though all DNA fragments are the same size (~215 base pairs), their rate of migration through the gel depends on the relative location of the A-tract. When the A-tract is located in the middle, the DNA fragment is more bent and migrates slowly; when the A-tract is near the end, the fragment is less bent and migrates faster. The lane marked MM contains molecular markers that provide a reference for DNA fragment size in base pairs (bp).
Regions of DNA where the two complementary strands have the same sequence when read in the 5′→3′ or the 3′→5′ direction occur relatively frequently in chromosomal DNA and are called palindromes (Figure 6-20a). In language, a palindrome is a word, phrase, or sentence that is spelled identically when read either forward or backward; two examples are ROTATOR and NURSES RUN. In biology, the term applies to double-stranded regions of DNA where one strand’s sequence is identical to its complement; for example, 5′-GAATTC-3′ is a palindrome because its complementary sequence is also 5′-GAATTC-3′. Inverted repeats are complementary sequences that occur on the same strand of a DNA or RNA molecule but in inverse directions, and often with some sequence separation between the repeats (Figure 6-20b). Such inverted repeats have the ability to form hairpin or cruciform (cross-shaped) structures through base pairing between the adjacent repeats. A related arrangement is a mirror repeat, in which the inverted repeat sequence is nonpalindromic (Figure 6-20c).
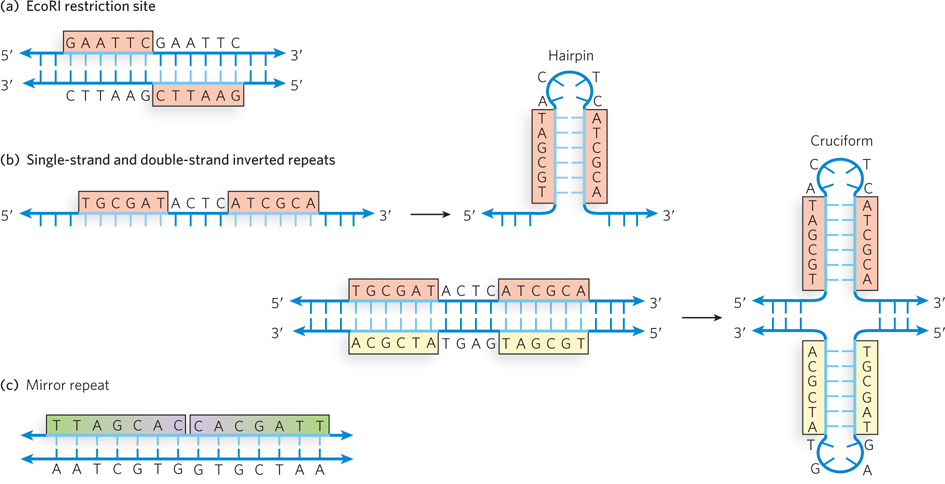
Figure 6-20: Palindromes, hairpins, cruciforms, and mirror repeats in DNA. (a) A palindromic sequence in DNA, such as the recognition site for the EcoRI restriction enzyme, is the same when reading from 5′ to 3′ on either strand of the DNA. (b) Inverted repeats within a single strand of DNA can be converted into a hairpin, which is double-stranded in the stem region. Inverted repeats within a double-stranded DNA sequence can form a cruciform (double-hairpin) structure. (c) DNA can also contain mirror repeats, such as TTAGCACCACGATT, which are not palindromic.
Palindromic sequences, inverted repeats, and mirror repeats play important biological roles, such as slowing or blocking protein synthesis by the ribosome—a process called translation attenuation (see Chapter 18)— or forming recognition sites for restriction enzymes, which catalyze double-stranded DNA cleavage (see Chapter 7). Inverted repeats, with their potential to form hairpins and cruciforms, are found in virtually every large DNA molecule and can encompass a few base pairs or thousands. The extent to which inverted repeats occur as cruciforms in cells is not known, but some cruciform structures have been demonstrated in vivo in Escherichia coli. Cruciforms are also known to form transiently during recombination of DNA molecules, in which genetic information on two chromosomes is exchanged during cell division. For this reason, palindromes can be involved in pathological chromosomal translocations, in which sequences from one chromosome are appended aberrantly to those of a nonhomologous chromosome, due to short segments of overlapping sequence between the two. The consequences of such events can be disastrous, leading to genetic dysregulation and cancer.
Several unusual DNA structures involve three or even four DNA strands. Although rare, these structural variants merit investigation because there is a tendency for them to form at sites where important events in DNA metabolism (replication, recombination, transcription) are initiated or regulated. Nucleotides participating in Watson-Crick base pairing have the potential to form additional hydrogen bonds, particularly with functional groups arrayed in the major groove. For example, a thymidine can pair with the adenosine of an A=T nucleotide pair, and a cytidine (if protonated) can pair with the guanosine of a G≡C pair, forming “base triples” (Figure 6-21a). The N-7, O6, and N6 of purines, the atoms that participate in this additional hydrogen bonding, are often referred to as Hoogsteen positions, and the non-Watson-Crick pairing is called Hoogsteen pairing. Karst Hoogsteen, in 1963, was the first to recognize the potential for these unusual pairings. Hoogsteen pairing allows the formation of triplex DNAs (Figure 6-21b).
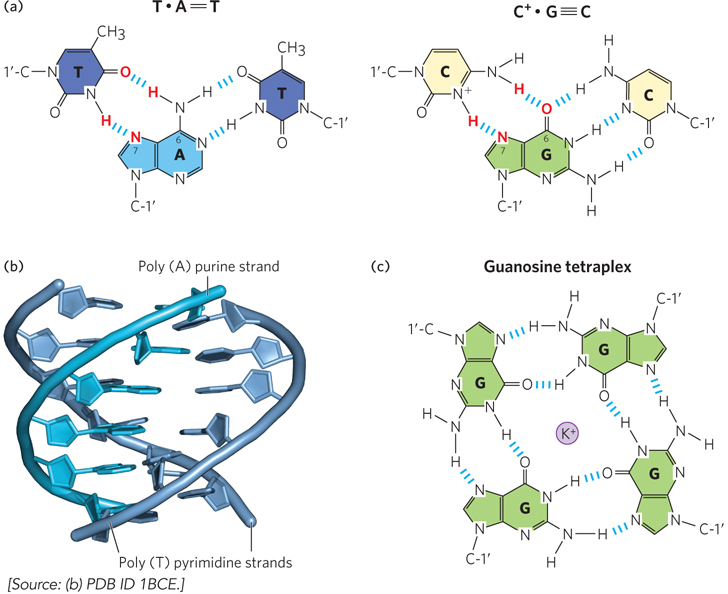
Figure 6-21: Three- and four-stranded DNA structures. (a) Base pairing in triplex DNA. Atoms participating in Hoogsteen pairing are in red; canonical Watson-Crick base pairs are in black. (b) Side view of a triple-helical DNA containing two poly(T) strands and one poly(A) strand. The dark blue and light blue strands in the foreground are antiparallel and engage in canonical Watson-Crick base pairing. The poly(T) strand in the background is parallel to the poly (A) strand and is paired through Hoogsteen hydrogen bonding. (c) One layer of a guanosine tetraplex (quadruplex) structure, showing hydrogen bonding of the bases. A K+ ion in the center of the tetraplex stabilizes the structure by coordinating the bases’ functional groups.
The triplexes form most readily within long sequences containing only pyrimidines or only purines in a given strand. Some triplex DNAs contain two pyrimidine strands and one purine strand; others contain two purine strands and one pyrimidine strand. DNA triplex formation can be highly sequence-specific. For example, triplex formation between a small section of chromosomal DNA and a chemically modified single-stranded DNA enabled Peter Dervan and his colleagues to cleave a human chromosome at a single site. They did this by synthesizing short oligonucleotides that could form triplex interactions with various segments of the chromosomal DNA. Because each oligonucleotide had a chemical modification at one end that triggered DNA strand breakage, the researchers were able to fragment the DNA at specific sites—which helped in mapping the location of the gene for the inherited neurological disorder known as Huntington disease.
Four DNA strands can also associate to form a tetraplex (or quadruplex), but this occurs readily only for DNA sequences with a very high proportion of G residues (Figure 6-21c). The guanosine tetraplex, or G tetraplex, is quite stable over a wide range of conditions. The DNA regions at the ends of linear chromosomes, called telomeres, typically consist of G-rich segments that have a propensity to form tetraplex structures when tested in the laboratory. Whether such structures contribute to the stability and recognition of telomeres in vivo is not known.
In the DNA of living cells, sites recognized by many sequence-specific DNA-binding proteins are arranged as palindromes, and polypyrimidine or polypurine sequences that can form triple helices are found within regions involved in regulating the expression of some eukaryotic genes. In principle, synthetic DNA strands designed to pair with these sequences to form triplex DNA could disrupt gene expression. This approach to controlling cellular metabolism is of growing commercial interest for its potential application in medicine and agriculture. Unusual DNA structures can also be engineered, raising the possibility of using DNA as a container to deliver drugs or proteins (Highlight 6-1).
HIGHLIGHT 6-1 TECHNOLOGY: DNA Nanotechnology
The ability of a single strand of DNA to base-pair specifically with its complementary sequence is essential for the accurate replication of encoded information. Scientists have also recognized that base pairing is an extremely useful property for assembling various three-dimensional structures from DNA. These observations have given rise to the field of DNA nanotechnology, the design and production of nucleic acid structures to be used for a range of technological applications. Instead of carrying genetic information, the nucleic acids used in this field of research are employed as engineering materials by taking advantage of their ability to base-pair and form various kinds of complex architectures. An example is shown in Figure 1 and is further described below.
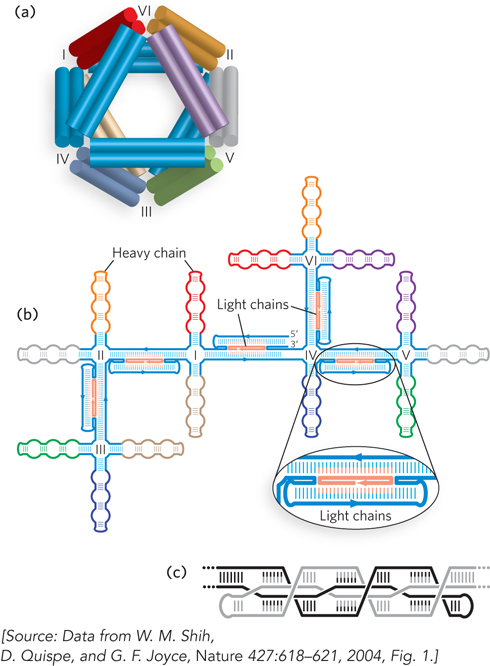
FIGURE 1 (a) A three-dimensional DNA octahedral structure as an example of DNA nanotechnology. The structure has 12 struts—the octahedron edges (double cylinders; each cylinder is a double-stranded DNA)—connected by six flexible joints (labeled I–VI). The joints are four-way junctions that connect the core-layer double helices of each strut. Colors correspond to the colored segments in (b), which shows the secondary structure of the branched-tree folding intermediate. This consists of a single 1,669-nucleotide DNA chain, or heavy chain, and five unique 40-nucleotide light chains (orange). (c) A close-up of the base-pairing scheme for each strut. Black and gray indicate two separate parts of the DNA strand that interact to form a strut. The cross-over base pairing gives the strut its structure and strength.
Based on concepts put forth by Nadrian Seeman in the 1980s, DNA nanotechnology has attracted increasing attention in recent years due to the ease and low cost of synthesizing DNA molecules of different lengths and sequences. The base-pairing rules for DNA, together with its inherent ability to form relatively rigid helical structures (both between and within DNA strands), enables the rational design of sequences that automatically assemble to form complex molecular architectures. For this reason, DNA nanotechnology building blocks—the DNA sequences used to build molecular devices—have been called “programmable matter.”

Gerald Joyce
DNA nanotechnology continues to advance toward various kinds of useful applications. For example, DNA devices might one day be the basis for “smart drugs” that allow targeted delivery of therapeutics. Scientists envision that a hollow DNA container encapsulating proteins capable of inducing cell death could be programmed to open only when in contact with cancerous cells. It might even be possible to produce these artificial structures in engineered living cells. This would then enable the targeted evolution of improved self-assembling DNA nanostructures.
The trick in bringing such concepts into being is to control the DNA base-pairing interactions to favor the formation of desired shapes. A particular challenge is to design sequences that can be cloned and copied by DNA polymerase enzymes in cells and that are also capable of self-assembling into specific three-dimensional shapes. Gerald Joyce and his colleagues at the Scripps Research Institute succeeded in designing a DNA sequence that could fold up into an octahedron in the presence of short complementary oligonucleotides. The research group produced a 1,669-nucleotide, single-stranded DNA molecule containing many such self-complementary sequences to enable specific base pairing between segments (see Figure 1). To induce three-dimensional folding, five 40-nucleotide oligonucleotides were added to form the struts of an octahedron by base pairing to distinct sites within the 1,669-nucleotide DNA. DNA mixtures were heated to disrupt base pairing and make single strands accessible, and then cooled to induce formation of base pairs. The changes in DNA structure resulting from base-pair formation were monitored by observing changes in the mobility of the DNA through a gel matrix and visualizing assembled DNA octahedra by electron microscopy (Figure 2). The microscopic analysis showed that, as expected from the sequence design, the DNA strands folded with 12 struts or edges joined at six four-way junctions to form hollow octahedra approximately 22 nm in diameter.

FIGURE 2 Gel electrophoresis and electron microscopy revealed the DNA octahedron assembly through base pairing. (a) Agarose gel electrophoresis of octahedron-forming DNA under different conditions. Lane MM: molecular marker lane with DNA size standards (number of base pairs (bp) is indicated on the left). Lane 1: 1,669-nucleotide strand (heavy chain) folded in the absence of Mg2+. Lane 2: 1,669-nucleotide strand folded in the presence of Mg2+. Lane 3: 1,669-nucleotide strand and 40-nucleotide light chains folded in the absence of Mg2+. Lane 4: 1,669-nucleotide strand and 40-nucleotide light chains folded in the presence of Mg2+. The Mg2+ shields the negative charges of the backbone phosphates and also promotes base pairing, creating a more compact structure that migrates more quickly through the gel. The resolution of the gel is not sufficient to detect differences in mobility of the DNA with or without the 40-nucleotide light chains. (b) Three views of the three-dimensional images of assembled DNA octahedra generated computationally, using electron micrographs as the starting point.
In theory, such DNA structures could be chemically modified to provide binding sites for small molecules or proteins within the enclosed space. If this idea can be shown to work, simple DNA base pairing will have been harnessed as a practical tool for drug delivery.
SECTION 6.2 SUMMARY
Chargaff’s rules state that in double-stranded DNA, the number of adenine nucleotides equals the number of thymine nucleotides (A=T), and the number of guanine nucleotides equals the number of cytosine nucleotides (G=C).
Using Franklin’s and Wilkins’s x-ray diffraction data from DNA fibers, Watson and Crick proposed that native DNA consists of two antiparallel chains in a right-handed double-helical arrangement. The hydrophilic sugar–phosphate backbone of each strand is on the outside of the helix, and the planar purine and pyrimidine bases project inward, perpendicular to the backbone axis. Complementary base pairs, A=T and G≡C, are formed by hydrogen bonding within the helix, consistent with Chargaff’s rules. The helical structure is further stabilized by shared electrons between the stacked planar base pairs.
In B-DNA, the most common form of DNA in cells, the base pairs are stacked nearly perpendicular to the long axis of the double helix, 3.4 Å apart, with 10.5 base pairs per turn (in solution).
Two other variations on DNA structure are A-DNA and Z-DNA. Like B-DNA, A-DNA is a right-handed helix, but it is more compact, with 11 base pairs per turn. A-DNA is favored in solutions that lack water, such as reagents used to crystallize DNA. Z-DNA forms a left-handed helix that contains 12 base pairs per turn and occurs only in sequences rich in C and G residues. Evidence suggests that eukaryotic DNA contains short stretches of Z-DNA, which might function in genetic recombination or the regulation of gene expression.
Repetitive sequences, such as tracts of A or T residues separated by G- or C-rich segments, cause bends in the DNA molecule. Bending can help facilitate DNA-protein binding.
DNA strands with inverted repeat sequences can form hairpin or cruciform structures that play roles in recombination and regulation of gene expression. Triplex or tetraplex forms of DNA can occur, though rarely, and may function in DNA metabolism.