6.4 CHEMICAL AND THERMODYNAMIC PROPERTIES OF NUCLEIC ACIDS
To understand how nucleic acids function, we must understand their chemical properties as well as their structures. The role of DNA as a repository of genetic information depends in part on its inherent stability. The chemical transformations that do happen are generally very slow in the absence of an enzyme catalyst. The long-
196
In addition to providing insights about physiological processes, our understanding of nucleic acid chemistry gives us a powerful array of technologies that have applications in molecular biology, medicine, and forensic science. We examine here the chemical properties of DNA and RNA, as well as some of these technologies. Many more techniques and applications are discussed in Chapter 7.
Double-Helical DNA and RNA Can Be Denatured
Solutions of carefully isolated, double-
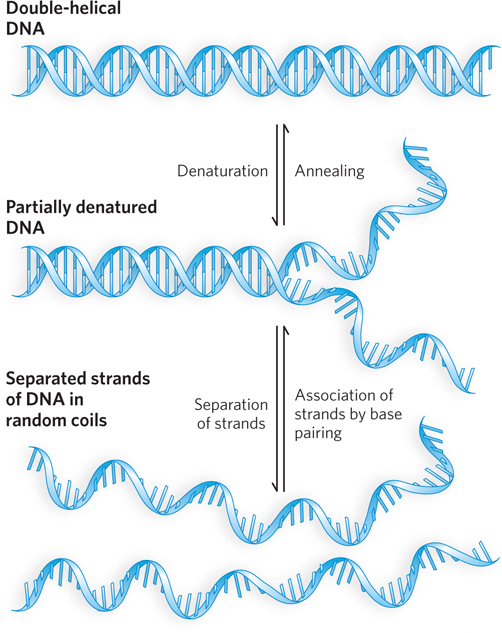
Renaturation of a DNA or RNA molecule is a rapid, one-
197
The close interaction of stacked bases in a nucleic acid has the effect of decreasing its absorption of UV light relative to that of a solution with the same concentration of free nucleotides, and the absorption is further decreased by the pairing of two complementary strands. Hydrogen bonding and base stacking in the double helix limit the resonance of the aromatic rings of the bases, thereby decreasing UV light absorption. This is known as the hypochromic effect (Figure 6-29). When DNA is denatured, the base pairs are disrupted and the two strands separate into randomly coiled chains. The resonance of the bases in each strand is no longer constrained, as it is when the bases are part of a double helix. As a result, the UV light absorption of single-
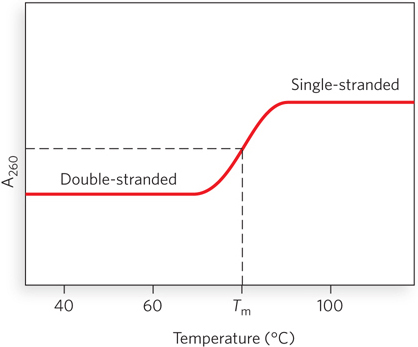
DNA molecules in solution denature when they are heated slowly. Each species of DNA has a characteristic denaturation temperature, or melting point (Tm), defined as the temperature at which half the DNA is denatured. In general, the higher the content of G≡C base pairs in the DNA, the higher its melting point. This is because G≡C base pairs, with three hydrogen bonds, require more heat energy to dissociate than do A=T base pairs. Careful determination of the melting point of a DNA specimen, under fixed conditions of pH and ionic strength, can yield an estimate of its base composition. If denaturation conditions are carefully controlled, regions that are rich in A=T base pairs will specifically denature while most of the DNA remains double-

RNA duplexes or RNA-
198
Nucleic Acids from Different Species Can Form Hybrids
The ability of two complementary DNA strands to pair with each other can be used to detect similar DNA sequences in different species or within the genome of a single species. If double-
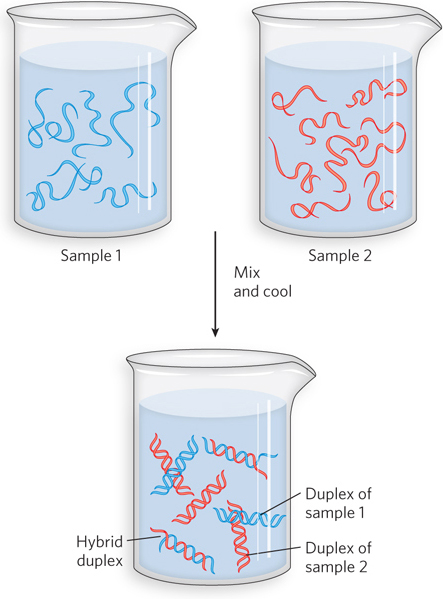
The hybridization of DNA strands from different sources forms the basis for powerful techniques used in classical molecular genetics. Although these hybridization techniques are used less often as new, high-
A specific gene’s DNA or RNA sequence can be detected in the presence of many other sequences by hybridization with a probe, a carefully chosen nucleic acid sequence complementary to the gene of interest. To be visualized in the laboratory, the probe must be labeled in some way, usually radioactively or with a fluorophore (a compound carrying a fluorescent group). The probe that is selected depends on what is known about the gene under investigation. Sometimes, a gene from another species that has sequence similarity to the gene of interest makes a suitable probe. If the protein product of a gene has been purified, probes can be designed and synthesized by working backward from the amino acid sequence, deducing the DNA sequence that would code for it. Or, researchers can often obtain the DNA sequence information necessary for creating a probe from sequence databases that detail the structure of millions of genes from a wide range of organisms. Because base-
199
In other common hybridization methods, gel electrophoresis is used to separate DNA or RNA molecules by size (Figure 6-32a). A variation of gel electrophoresis, used to detect proteins under denaturing conditions, was discussed in Chapter 4 (see Highlight 4-1). Here, the gel matrix is not denaturing but instead is made of agarose, a kelp-
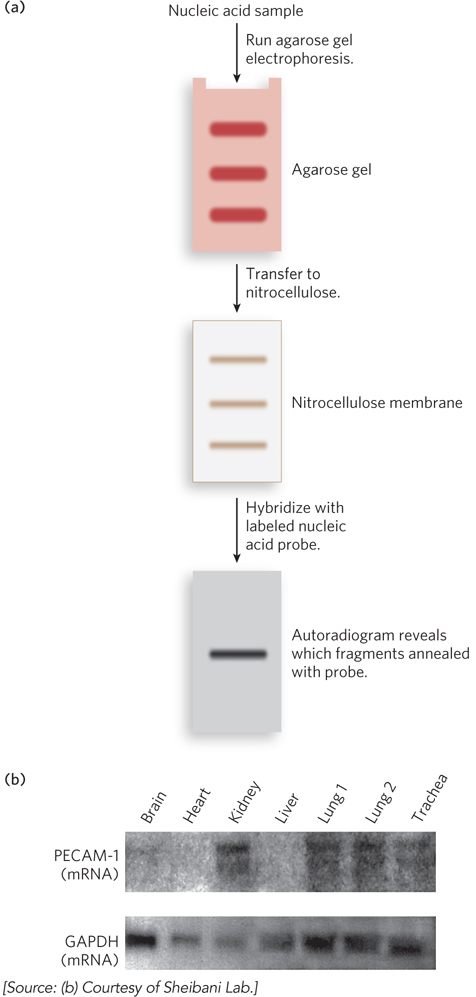
When used to detect DNA, this method is known as Southern blotting, named for Edwin Southern, who invented the technique at the University of Edinburgh. When used for detecting RNA, the technique is called Northern blotting, because of its similarity to the Southern method. Applications of these techniques include identifying a person on the basis of a single hair left at the scene of a crime or predicting the onset of a disease in an individual decades before symptoms appear. Northern blotting can also be used to detect the levels of a particular type of RNA in different body tissues (Figure 6-32b), providing fascinating insight into how cells regulate the expression of genes. Notably, these classical methods of Southern and Northern blotting are still used to answer specific experimental questions, despite the development of high-
Nucleotides and Nucleic Acids Undergo Uncatalyzed Chemical Transformations
Purines and pyrimidines, and the nucleotides of which they are a part, can undergo spontaneous alterations in their covalent structure. The rate of these reactions is generally very slow, but as noted earlier, they are physiologically significant because of the cell’s low tolerance for changes in its genetic information. Alterations in DNA structure that produce permanent genetic changes are known as mutations. Extensive evidence suggests an intimate link between the accumulation of mutations in an individual organism and the processes of aging and carcinogenesis.
200
Several nucleotide bases undergo deamination, a spontaneous loss of their exocyclic amino groups. For example, under typical cellular conditions, deamination of cytosine (in DNA) to uracil occurs in about 1 of every 107 C residues in 24 hours (Figure 6-33). This corresponds to about 100 spontaneous deamination events per day, on average, in a mammalian cell. Deamination of adenine and guanine occurs at about 1/100th this rate.
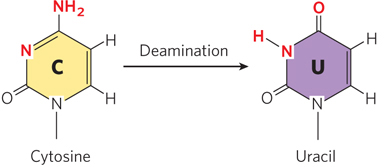
The slow cytosine deamination reaction seems innocuous enough, but it is almost certainly the reason that DNA contains thymine rather than uracil. In DNA, uracil is the product of cytosine deamination, and it is readily recognized as foreign and is removed by a DNA repair system (see Chapter 12). If DNA normally contained uracil, the recognition of U residues resulting from cytosine deamination would be more difficult, and unrepaired uracils would lead to permanent sequence changes as they were paired with adenines during replication (cytosine normally pairs with guanine, so introduction of uracil into DNA effectively changes a C≡G base pair to a U–
Cytosine deamination also provides innate cellular defense against viral infection. A family of human proteins called APOBECs catalyze cytosine deamination in the viral genome during the initial round of replication by HIV. This hypermutation results in many nonviable viral particles, eventually destroying the coding capacity of the virus. In HIV and related viruses, the viral protein Vif binds to APOBECs and triggers their degradation. Vif has therefore become an important antiviral target, because viruses lacking this protein are much less capable of establishing chronic infection in human cells.
Base Methylation in DNA Plays an Important Role in Regulating Gene Expression
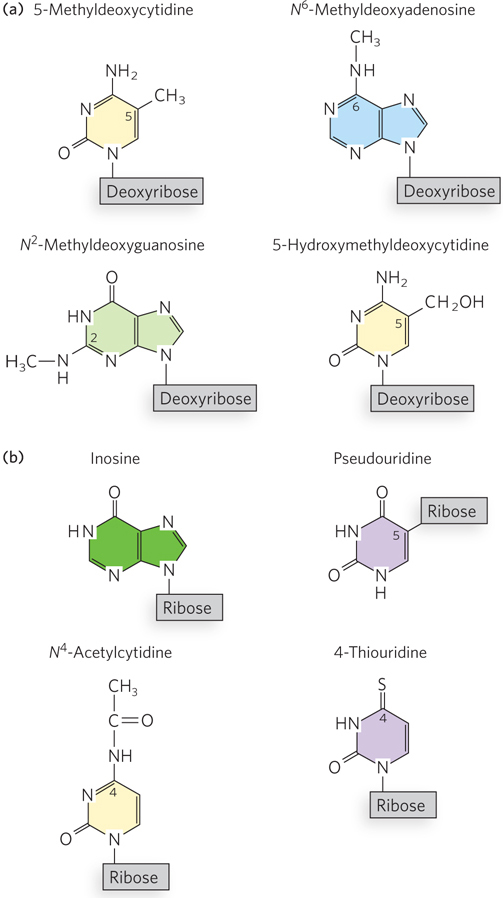
Certain nucleotide bases in DNA molecules are enzymatically methylated, usually after DNA synthesis is complete. Adenine and cytosine are methylated more frequently than guanine (Figure 6-34a). Methylation is generally confined to certain sequences or regions of a DNA molecule. For example, more than half of all CpG sequences in mammalian genomes are methylated on the C residue. Methylation tends to inhibit gene expression, because the methylated DNA is not efficiently copied into RNA. In many cancers, gene regulatory regions in DNA become abnormally hypermethylated. This can result in the silencing of genes that would otherwise control cell growth. DNA methylation may affect gene transcription by physically blocking the binding of proteins that facilitate transcription. Other proteins, however, can specifically bind to methylated DNA and recruit additional proteins that help form compact, inactive regions of chromosomal DNA.
201
All known DNA methylases (methyltransferases) use S-adenosylmethionine as a methyl group donor (see Figure 6-13). E. coli has two prominent methylation systems. One serves as part of a defense mechanism that helps the cell distinguish its DNA from foreign DNA by marking its own DNA with methyl groups and destroys foreign, nonmethylated DNA, a process known as restriction modification (discussed in Chapter 7). The other system methylates A residues in the sequence 5′-GATC-
In eukaryotic cells, about 5% of all C residues in DNA are methylated to form 5-
KEY CONVENTION
When a chemical group attached to an atom in the purine or pyrimidine ring is altered, the ring position of the substituent is indicated by the number of that atom—
RNA Molecules Are Often Site-Specifically Modified In Vivo
Like DNA, many functional RNAs are posttranscriptionally modified at specific nucleotides (Figure 6-34b). Some of the first examples were discovered in ribosomal and transfer RNAs. In some cases, modifications involve the addition of a functional group to an existing nucleotide in the sequence. For example, a methyl group can be added to the 2′ hydroxyl of ribose, thereby blocking its ability to form a hydrogen bond. In bacteria, some C residues of tRNAs are modified to N4-acetylcytidine in a process thought to contribute to the accuracy of protein synthesis. In other cases, the base itself is changed, or its linkage to the ribose—
Many of the enzymes that catalyze these chemical modifications of RNA have been identified. They are often evolutionarily conserved, indicating that RNA modification has been occurring in biological systems for a long time. More difficult to figure out is the function of these chemical changes in RNA. Molecular biologists can produce unmodified versions of RNAs in the laboratory and compare their functions with those of the chemically altered counterparts isolated from cells. This approach has only rarely discerned much of an effect of a modified base. However, genetic experiments in which an RNA-
The Chemical Synthesis of DNA and RNA Has Been Automated
Knowledge of DNA and RNA chemistry provided the basis for devising methods to synthesize nucleic acids in the laboratory. This technology has paved the way for many biochemical advances that depend on the ability to synthesize oligonucleotides with any chosen sequence. The chemical methods for synthesizing nucleic acids were developed primarily by H. Gobind Khorana and his colleagues in the 1970s. Refinement and automation of these methods have made possible the rapid and accurate synthesis of DNA strands.
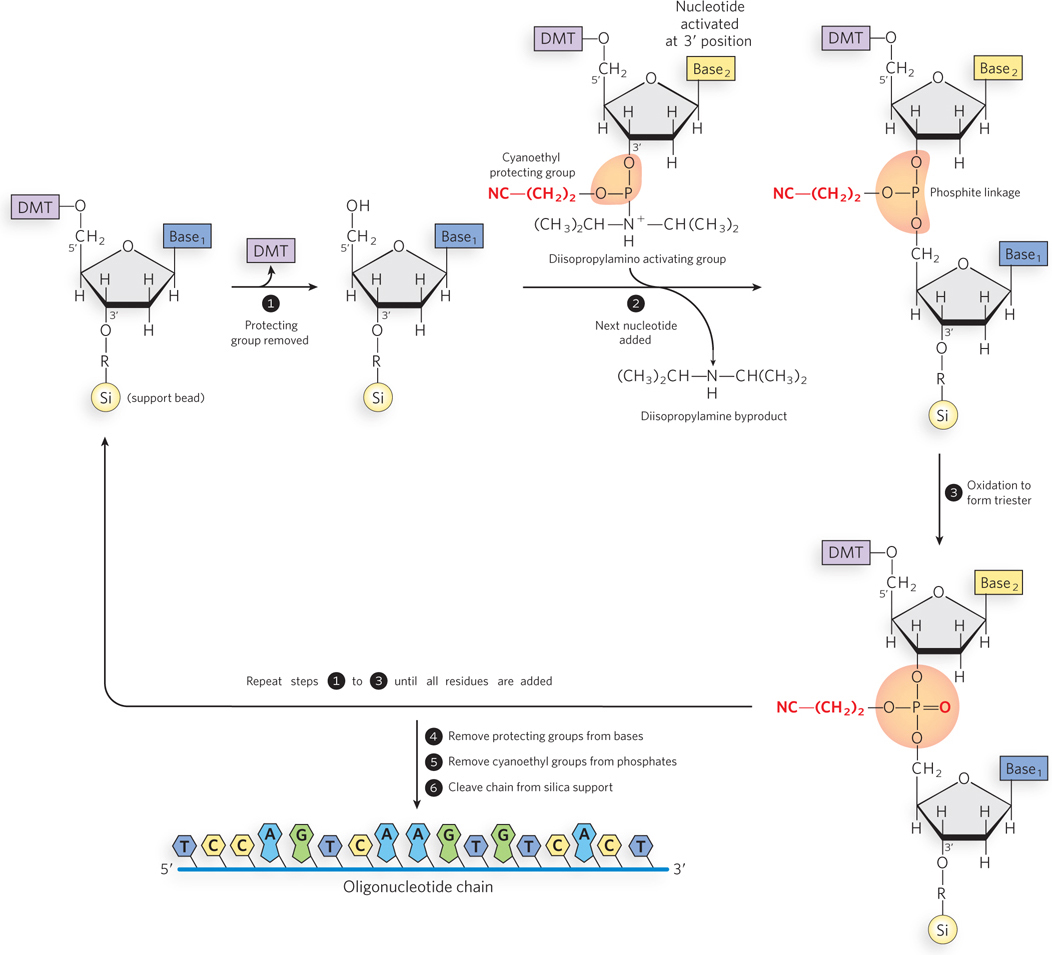
DNA (or RNA) synthesis is carried out with the growing strand attached to a solid support (Figure 6-35). First, a nucleotide is attached to the support, a glass or polystyrene bead, through its 3′-hydroxyl group, and polynucleotide synthesis proceeds in the 3′→5′ direction. This is the opposite of the direction of biological polynucleotide synthesis by polymerase enzymes, which is 5′→3′. Functional groups on the bases and phosphates, including hydroxyl and amine groups, are transiently protected with chemical groups that are readily removed after synthesis is complete. The 5′-hydroxyl group is temporarily protected by a dimethoxytrityl (DMT) group; the DMT group is removed from the end of the growing polymer chain at the beginning of each cycle (step 1 in Figure 6-35) to permit extension of the chain by another nucleotide (step 2). Oxidation of the phosphite linkage between the nucleotides completes the cycle (step 3). When chain synthesis is complete, protecting groups are removed from the bases and phosphates, and the oligonucleotide chain is cleaved from its solid support (steps 4, 5, 6). The efficiency of each addition step is very high, allowing the routine laboratory synthesis of polymers containing 70 to 80 nucleotides and, in some laboratories, much longer strands.
202
Oligonucleotide synthesis is very useful for techniques such as Southern and Northern blotting and for the polymerase chain reaction (PCR) and DNA sequencing, which are discussed in Chapter 7. In addition, chemical synthesis makes it possible to incorporate chemical modifications in the polymer product, such as biotin groups, extra phosphates, sulfhydryl groups, and methyl groups. These functional groups are useful for such applications as specific labeling of a DNA strand or stabilization of an RNA oligonucleotide against enzymatic degradation in cells.
203
SECTION 6.4 SUMMARY
Native DNA undergoes reversible unwinding and separation of the strands (melting) on heating or at extremes of pH. DNAs rich in G≡C base pairs have higher melting points than DNAs rich in A = T base pairs.
Hybridization, or base pairing between two strands of nucleic acid from different sources, is the basis for important techniques used to study and isolate specific genes and RNAs.
Southern blotting is a method by which a specific DNA sequence can be identified in a mixture, following size-
based fractionation of the DNA sample by agarose gel electrophoresis. A probe complementary to the DNA of interest is labeled with a radioactive or fluorescent functional group. After transferring the size- fractionated DNA from the gel to a membrane, the probe is hybridized to the sample on the membrane so that the sequence of interest can be visualized. Northern blotting, analogous to Southern blotting, is used for detecting specific RNA sequences.
Mutations are alterations in DNA structure that produce permanent changes in the encoded genetic information. Deamination of cytosine is a common chemical mutation in DNA that can damage the genetic code if not corrected by the cell. Deamination of viral nucleic acid can be used to defend against viral infection.
Certain A and C residues in DNA are often enzymatically methylated after DNA synthesis. E. coli uses methylation to distinguish between host and foreign DNA and to facilitate the repair of mismatched base pairs that arise from replication errors. In eukaryotes, DNA methylation often inhibits gene expression.
Some residues in RNAs are chemically modified by enzymes that introduce methyl or acetyl groups at specific sites or alter a nucleotide base in other ways. These modifications may stabilize RNA structures and can also influence RNA recognition by proteins.
DNA and RNA polymers of any sequence can be synthesized with simple, automated procedures involving chemical and enzymatic methods. Solid-
phase synthesis of DNA and RNA occurs in the 3′→5′ direction, using chemically protected nucleotides that are selectively deprotected and coupled to the growing polynucleotide chain in successive cycles.
UNANSWERED QUESTIONS
Although many details of nucleic acid structure are well understood, future challenges involve linking the chemistry of these molecules to their behavior in biological systems. Here are several interesting questions in the field.
What are the functions of noncanonical DNA structures in cells? We do not yet know whether non-
B- DNA functions in specific cellular processes. For example, some evidence suggests that with its left- handed twist, Z- DNA relieves some of the torsional strain that would otherwise build up during DNA transcription. Perhaps for this reason, the potential to form Z- DNA structures correlates with genomic regions of active transcription. But definitive proof of these ideas has been elusive. Whether three- stranded or four- stranded structures are biologically relevant is also a topic that remains ripe for experimentation. Do mRNAs have stable three-
dimensional structures? Although mRNAs were once thought to be spaghetti- like molecules, increasing evidence hints that they may have stable structures that contribute to biological function. For example, many mRNAs include long sequences that extend beyond the coding region of the gene and are critical for proper gene regulation. Specific proteins bind to these regions and probably recognize structures within them. How widespread is chemical modification of RNA? Modified nucleotides in tRNA and rRNA have been recognized for a long time, but we do not know whether other RNAs in cells contain such chemical changes. This is an important question, because modifications could influence the function of RNAs that play various roles in controlling gene expression and therefore might be relevant to understanding disease pathways.
204
HOW WE KNOW: DNA Is a Double Helix
Sayre, A. 1975. Rosalind Franklin and DNA. New York: W.W. Norton & Co.
Watson, J.D. 1968. The Double Helix: A Personal Account of the Discovery of the Structure of DNA. New York: Atheneum.
Watson, J.D., and F.H. Crick. 1953. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 171:737–
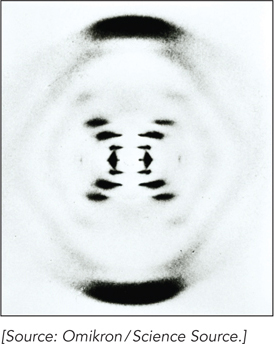
By the early 1950s, DNA had been confirmed as the genetic material in cells, but its structure was unknown. Given that structural information would be key to understanding heredity, the race was on to solve the mystery of DNA structure. Researchers including Rosalind Franklin, Raymond Gosling, and Maurice Wilkins were measuring x-
Rosalind Franklin’s famous Photograph 51 revealed a particularly well-

Watson and Crick’s work was published in a letter to the British journal Nature in 1953. In the same issue, several other papers provided experimental support for the Watson-
205
DNA Helices Have Unique Geometries That Depend on Their Sequence
Wing, R., H. Drew, T. Takano, C. Broka, S. Tanaka, K. Itakura, and R.E. Dickerson. 1980. Crystal structure analysis of a complete turn of B-
The discovery of the DNA double helix marked the dawn of molecular biology. However, it was not until 1980 that the first single crystal of a DNA molecule was obtained. This was an important landmark in its own right, because for the first time it became possible to determine the exact helical parameters of a defined DNA sequence. Why did it take almost 30 years after the work of Watson, Crick, Franklin, Wilkins, and Gosling for specific DNA sequences to be crystallized?
The answer is technology. Until the late 1970s, it wasn’t possible to synthesize DNA molecules in the laboratory, so investigators could not produce enough of a specific sequence to make growth of single crystals feasible. Once the methodology was available to synthesize DNA oligonucleotides on solid supports, short DNA molecules of specific length and sequence could be produced in milligram quantities. This material could be purified, and it crystallized readily when concentrated slowly in the presence of suitable buffers. Single crystals of DNA offered some distinct advantages over the DNA fibers analyzed by Franklin, Gosling, and Wilkins. DNA fibers can readily form from a mixture of DNAs of different lengths and sequences, and the structures obtained by analyzing the fiber diffraction patterns produce an “averaged” structure of all the molecules in the fiber. In contrast, single crystals, by definition, are formed by arrays of identical molecules.
Richard Dickerson and his colleagues recognized the wealth of information to be gained by solving a molecular structure of single DNA crystals. They used a self-
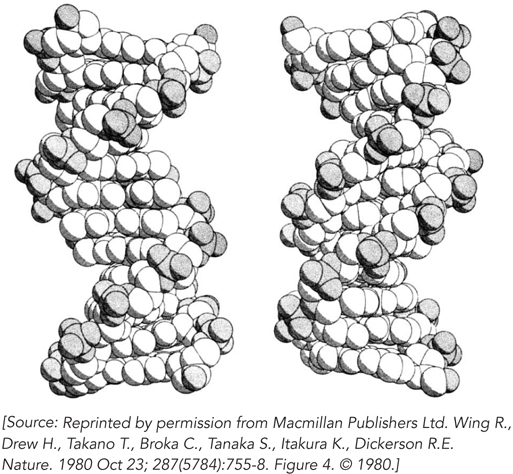
206
HOW WE KNOW: Ribosomal RNA Sequence Comparisons Provided the First Hints of the Structural Richness of RNA
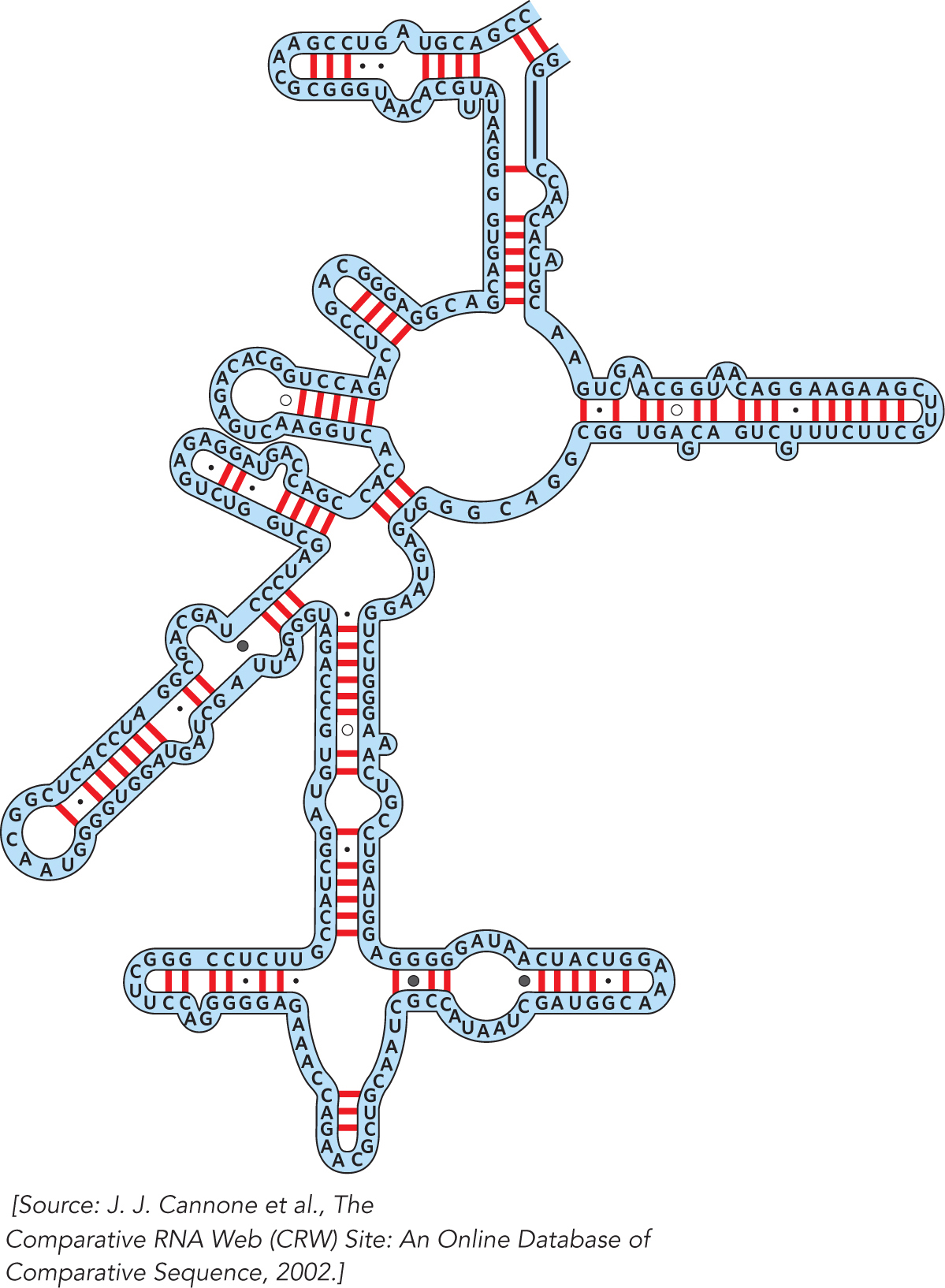
Gutell, R.R., N. Larsen, and C.R. Woese. 1994. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol. Rev. 58:10–
Ribosomes have been around for a long time, and the sequences of the RNAs they contain have been constrained over the course of evolution by the requirements of making functional ribosomes to catalyze protein synthesis. Carl Woese recognized in the 1960s that comparing ribosomal RNA sequences would provide valuable information about the evolutionary relationships among different organisms. Working over many years, he and his colleague Harry Noller assembled careful alignments of the 16S and 23S rRNAs from a large number of microbes. This work led Woese to propose the three-
The comparative analysis approach begun by Woese and Noller was continued by Robin Gutell, who expanded the comparison to include 16S and 23S rRNA sequences from multicellular organisms, including humans. Gutell’s critical analysis provided the first hints that these RNAs form specific three-
207