Folding of Proteins in Vivo Is Promoted by Chaperones
The conditions under which a purified, denatured protein refolds in a test tube differ markedly from the conditions under which a newly synthesized polypeptide folds in a cell. The presence of other biomolecules, some of which are themselves nascent and in the process of folding, can potentially interfere with the autonomous, spontaneous folding of an otherwise natively well-ordered protein by forming aggregates. The cytosolic concentrations of some proteins are very high, and the total cytosolic protein concentration can be ~300 mg/ml in mammalian cells. These high protein concentrations favor aggregate formation by increasing the chances a nascent protein will encounter other proteins prior to completing its folding. Unfolded and partly folded proteins tend to aggregate into large, often water-insoluble masses, from which it is extremely difficult for a protein to dissociate and then fold into its proper conformation. In part, this aggregation is due to the exposure of hydrophobic side chains that have not yet had a chance to be buried in the inner core of the folded protein. Exposed hydrophobic side chains on different molecules will stick to one another, owing to the hydrophobic effect (see Chapter 2), and thus promote aggregation. The risk of such aggregation is especially high for newly synthesized proteins that have not yet completed their proper folding. Intrinsically disordered proteins are much less likely to form aggregates because, at least in some cases, they have relatively fewer hydrophobic side chains that can mediate such aggregation. Although protein folding into a well-ordered native state can occur in vitro, this does not happen for all unfolded molecules in a timely fashion because of the very large number of potentially incorrect, intermediate conformations into which the protein might fold.
Page 83
Given such impediments, cells require faster, more efficient mechanisms for folding natively well-ordered proteins into their correct shapes than sequence alone provides. Without such help, cells might waste much energy in the synthesis of improperly folded, nonfunctional proteins, which would have to be destroyed to prevent their disrupting cell function. Cells clearly have such mechanisms, since more than 95 percent of the proteins present within cells have been shown to be in their native conformations. Proteins that do not or cannot fold properly—for example, those encoded by genes with mutations that alter the amino acid sequence—are often recognized as unfolded and rapidly degraded (hydrolyzed) by enzymes. The explanation for the cell’s remarkable efficiency in promoting proper protein folding is that cells make a set of proteins, called chaperones, that facilitate proper folding of nascent proteins. One way chaperones facilitate proper folding is to prevent aggregation by binding to the target polypeptide or sequestering it from other partially or fully unfolded proteins, thus giving the nascent protein time to fold properly. The importance of chaperones is highlighted by the observation that many are evolutionarily conserved. Chaperones are found in all organisms from bacteria to humans, and some are homologs with high sequence similarity that use almost identical mechanisms to assist protein folding.
Chaperones can fold newly made proteins into functional conformations, refold misfolded or unfolded proteins into functional conformations, disassemble potentially toxic protein aggregates that form due to protein misfolding, assemble and dismantle large multiprotein complexes, and mediate transformations between inactive and active forms of some proteins. Chaperones, which in eukaryotes are located in every cellular compartment and organelle, bind to the target proteins—also called substrates or client proteins—whose folding they will assist. Chaperones use a cycle of ATP binding, ATP hydrolysis to ADP, and exchange of a new ATP molecule for the ADP to induce a series of conformational changes that are essential for their function. There are several different classes of chaperones with distinct structures, all of which use ATP binding and hydrolysis in a variety of ways, which include (1) enhancing the binding of the target protein and (2) switching their own conformation. This ATP-dependent conformational switching is used (1) to optimize folding, (2) to return the chaperone to its initial state so that it is available to help fold another molecule, and (3) to set the time permitted for refolding, which can be determined by the rate of ATP hydrolysis.
Two general families of chaperones have been identified:
Molecular chaperones, which bind to a short segment of a protein substrate and stabilize unfolded or partly folded proteins, thereby preventing these proteins from aggregating and being degraded.
Chaperonins, which form folding chambers into which all or part of an unfolded protein can be sequestered, giving it time and an appropriate environment to fold properly.
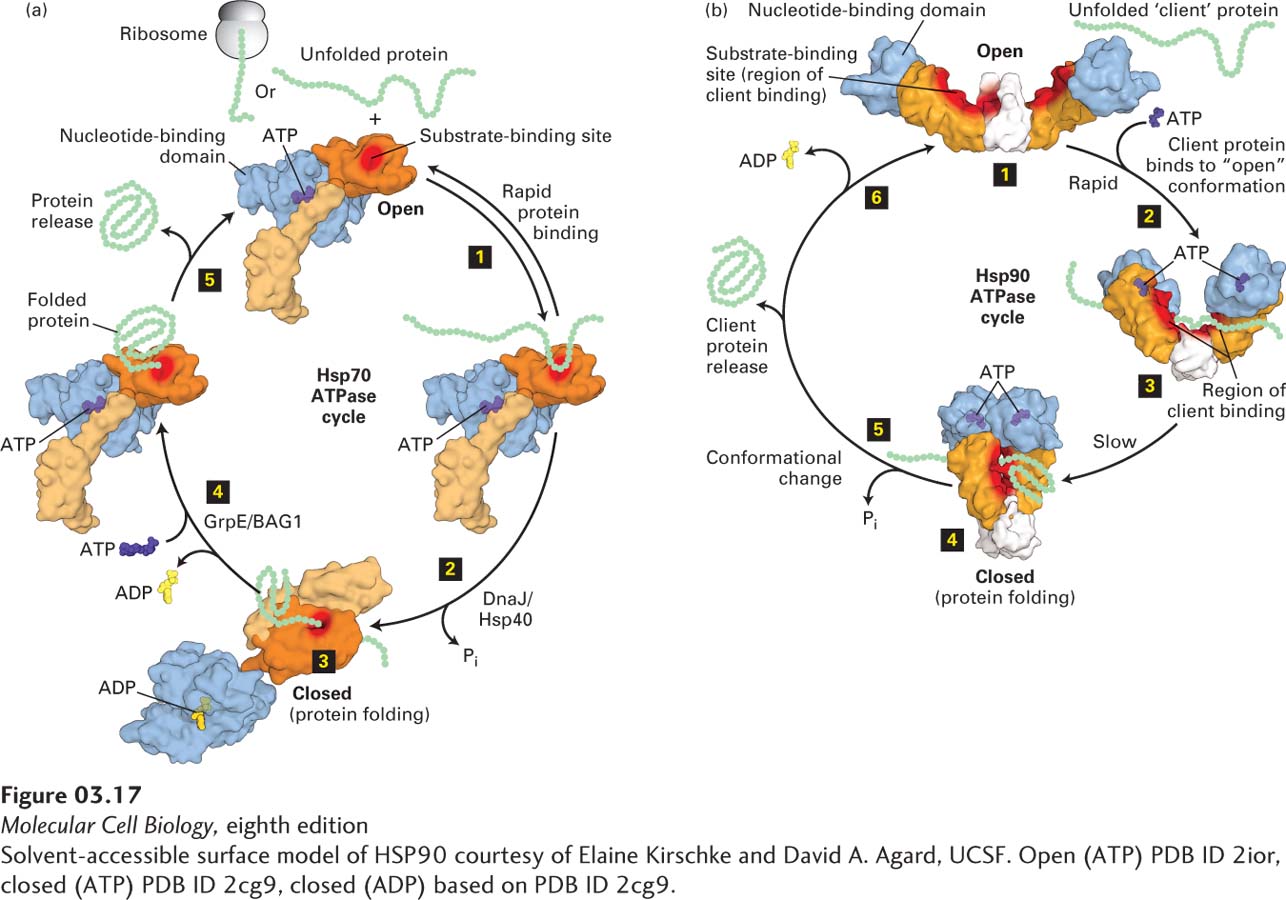
Molecular Chaperones The heat-shock protein Hsp70 in the cytosol and its homologs (Hsp70 in the mitochondrial matrix, BiP in the endoplasmic reticulum, and DnaK in bacteria) are molecular chaperones. They were first identified by their rapid appearance after a cell had been stressed by heat shock (Hsp stands for “heat-shock protein”). Hsp70 and its homologs are the major chaperones in all organisms that use an ATP-dependent cycle to fold their substrates (Figure 3-17a). When bound to ATP, the monomeric Hsp70 protein assumes an open conformation, in which an exposed hydrophobic substrate-binding pocket transiently binds to exposed hydrophobic regions of an incompletely folded or partially denatured target protein, and then rapidly releases this substrate, as long as ATP is bound (step 1 in Figure 3-17a). Hydrolysis of the bound ATP causes the molecular chaperone to assume a closed form that binds its substrate protein much more tightly, and this tighter binding appears to facilitate the target protein’s folding, in part by preventing it from aggregating with other unfolded proteins (step 2 in Figure 3-17a). Next the exchange of ATP for the chaperone-bound ADP (step 3) causes a conformational change in the chaperone that releases the target protein and regenerates an “empty,” ATP-bound Hsp70 ready to help fold another protein (step 4). If the target is now properly folded, it cannot rebind to an Hsp70. If it remains at least partially unfolded, it can bind again to give a chaperone another chance to help fold it properly. As we will see later in this chapter, a variety of proteins use a cycle of trinucleotide hydrolysis to a dinucleotide, followed by dinucleotide/trinucleotide exchange, to control their activities. Later in this chapter, we will discuss a group of proteins called GTPases that depend on the exchange of GTP, rather than ATP, for bound GDP (instead of ADP) to induce conformational changes that dramatically influence the proteins’ activities and the subsequent hydrolysis of the bound GTP to GDP.
[Solvent-accessible surface model of HSP90 courtesy of Elaine Kirschke and David A. Agard, UCSF. Open (ATP) PDB ID 2ior, closed (ATP) PDB ID 2cg9, closed (ADP) based on PDB ID 2cg9.]
FIGURE 3-17Molecular chaperone–mediated protein folding. (a) Hsp70. Many proteins fold into their proper three-dimensional structures with the assistance of Hsp70 or one of several Hsp70-like proteins. These molecular chaperones transiently bind to a nascent polypeptide as it emerges from a ribosome or to a protein that has otherwise unfolded. In the Hsp70 cycle, an unfolded substrate protein binds in rapid equilibrium (step 1) to Hsp70’s substrate-binding site (red) in the open conformation of its substrate-binding domain (light and dark orange) when an ATP (purple) is bound at Hsp70’s nucleotide-binding domain (light blue). The substrate-binding domain comprises two subdomains (light and dark orange) that change relative positions and conformations during the cycle. Co-chaperone accessory proteins (DnaJ/Hsp40) stimulate the hydrolysis of ATP to ADP (yellow) that induces a large conformational change in the substrate-binding domain, resulting in the closed conformation, in which the substrate is locked into the substrate-binding domain; here proper folding is facilitated (steps 2 and 3). Exchange of ATP for the bound ADP, stimulated by other accessory co-chaperone proteins (GrpE/BAG1), converts the Hsp70 back to the open conformation (step 4), releasing the properly folded substrate (step 5) and regenerating the open conformation, which can then interact with additional substrates. (b) Three conformational states of the dimeric Hsp90 molecular chaperone thought to be involved in substrate (also called client) remodeling. Client proteins bind at the substrate-binding site (red surface) shared by the substrate-binding (orange) and C-terminal dimerization (white) domains and are thought to be remodeled in response to ATP binding and hydrolysis. The Hsp90 cycle begins when there is no nucleotide bound to the nucleotide-binding domains (light blue) and the dimer is in a very flexible, open configuration (step 1) that can bind a client. Rapid ATP binding leads to a conformational change (step 2) in which the nucleotide-binding domains and the substrate-binding domains move together (intermediate shown in step 3) into a closed conformation in which the nucleotide-binding domains are dimerized (step 4). The precise locations in Hsp90 at which clients bind apparently vary for different clients, but the binding surface, including the intersection of the substrate-binding domains and C-terminal dimerization domains (highlighted by red shading) binds a number of clients. ATP hydrolysis results in a conformational change in Hsp90 (step 5) that may include a highly compact form, folding of the client, and client protein release. The ADP-bound form of Hsp90 can adopt several conformations, including a highly compact form. Release of ADP (step 6) regenerates the initial flexible open state, which can then interact with additional clients. See E. D. Kirschke et al., 2014, Cell157:1685 and M. Taipale, D. F. Jarosz, and S. Lindquist, 2010, Nat. Rev. Mol. Cell Biol.11:515.
[Solvent-accessible surface model of HSP90 courtesy of Elaine Kirschke and David A. Agard, UCSF. Open (ATP) PDB ID 2ior, closed (ATP) PDB ID 2cg9, closed (ADP) based on PDB ID 2cg9.]
Page 84
Additional proteins, such as the co-chaperone Hsp40 in eukaryotes (DnaJ in bacteria), help increase the efficiency of the Hsp70-mediated folding of many proteins not only by stimulating the binding of substrate, but also by increasing the rate of hydrolysis of ATP by 100- to 1000-fold (see step 2 in Figure 3-17a). Members of four different families of nucleotide exchange factors (e.g., GrpE in bacteria; BAG, HspBP, and Hsp110 in eukaryotes) also interact with Hsp70 (or DnaK), promoting the exchange of ATP for ADP (see step 3). Multiple molecular chaperones are thought to bind to all nascent polypeptide chains as they are being synthesized on ribosomes. In bacteria, 85 percent of the proteins are released from their chaperones and proceed to fold normally; an even higher percentage of proteins in eukaryotes follow this pathway.
Page 85
The Hsp70 protein family is not the only class of molecular chaperones. Another distinct class of molecular chaperones is the Hsp90 family, whose members usually recognize partially folded substrate proteins. Evolutionarily related Hsp90 family members are present in all organisms except archaea. Their strong evolutionary conservation is seen in the high amino acid sequence similarity (55 percent) of the Hsp90 from the bacterium E. coli and human Hsp90. In most eukaryotes, there are four distinct Hsp90s, two of which are in the cytosol (at 1–2 percent of total protein, Hsp90 is one of the most abundant cytosolic proteins) and one each in the endoplasmic reticulum and the mitochondrion. Although the range of protein substrates for Hsp90 chaperones is not as broad as for some other chaperones (at least 10 percent of yeast proteins are thought to be Hsp90 substrates), the Hsp90s are essential in eukaryotes. The Hsp90s help cells cope with denatured proteins generated by stress (e.g., heat shock), and they ensure that some of their substrates, usually called “clients,” can be converted from an inactive to an active state or otherwise held in a functional conformation. In some cases, an Hsp90 forms a relatively stable complex with a client until an appropriate signal causes its dissociation from the client, freeing the client to perform some regulated function in the cell. Hsp90 clients include transcription factors such as the receptors for the steroid hormones estrogen and testosterone. These steroid receptors regulate sexual development and function by controlling the activities of many genes (see Chapter 9). Another type of Hsp90 client is the set of enzymes called kinases, which control the activities of many proteins by phosphorylation (see Chapters 15 and 16).
Unlike monomeric Hsp70, Hsp90 functions as a dimer in a cycle in which ATP binding, hydrolysis, and ADP release are coupled to major conformational changes and to binding, folding or activation, and release of clients (Figure 3-17b). Although much about the mechanism of Hsp90 remains to be learned, it is clear that clients bind to the substrate-binding domains when the chaperone is in the “open” conformation (step 1 in Figure 3-17b), that ATP binding leads to interaction of the ATP-binding domains and formation of a “closed” conformation (steps 1 and 2 in Figure 3-17b), and that hydrolysis of ATP plays an important role in activation of some client proteins and their subsequent release from the Hsp90 (step 3). We also know that there are at least 20 co-chaperones that can have profound effects on the activity of Hsp90, including modulating its ATPase activity and determining which proteins will be clients (client specificity). Co-chaperones can also help coordinate the activities of Hsp90 and Hsp70. For example, Hsp70 can help begin the folding of a client that is then handed off by a co-chaperone to Hsp90 for additionalprocessing. Hsp90 activity can also be influenced by its covalent modification by small molecules. Finally, Hsp90 can help cells recognize misfolded proteins that are unable to refold and facilitate their degradation by mechanisms discussed later in this chapter. Thus, as part of the quality-control system in cells, molecular chaperones can help properly fold proteins or facilitate the destruction of those that cannot fold properly.
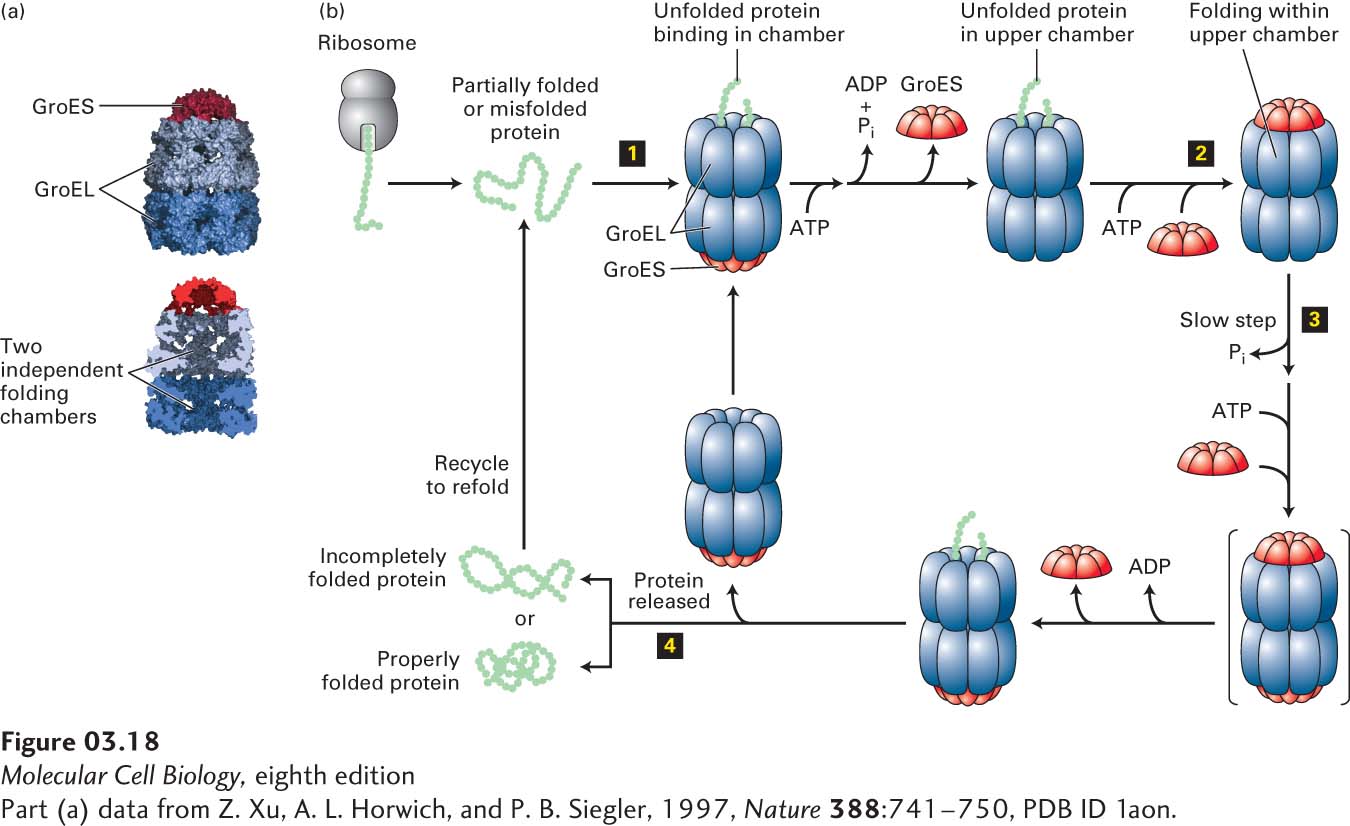
Chaperonins The proper folding of a large variety of newly synthesized proteins also requires the assistance of another class of proteins, the chaperonins, also called Hsp60s. These huge cylindrical supramolecular assemblies are formed from two rings of oligomers. There are two distinct groups of chaperonins that differ somewhat in their structures, detailed molecular mechanisms, and locations. Group I chaperonins, found in prokaryotes, chloroplasts, and mitochondria, are composed of two rings, each having seven subunits that interact with a homoheptameric co-chaperone “lid.” The bacteria group I chaperonin, known as GroEL/GroES, is shown in Figure 3-18a. In the bacterium E. coli, GroEL is thought to participate in the folding of about 10 percent of all proteins. Group II chaperonins, which are found in the cytosol of eukaryotic cells (e.g., TriC in mammals) and in archaea, can have eight to nine either homomeric or heteromeric subunits in each ring, and the “lid” function is incorporated into those subunits themselves—no separate lid protein is needed. It appears that ATP hydrolysis triggers the closing of the lid of group II chaperonins.
[Part (a) data from Z. Xu, A. L. Horwich, and P. B. Siegler, 1997, Nature388:741–750, PDB ID 1aon.]
FIGURE 3-18Chaperonin-mediated protein folding. Proper folding of some proteins depends on chaperonins such as the prokaryotic group I chaperonin GroEL. (a) GroEL is a barrel-shaped complex of fourteen identical ~60,000-MW subunits, arranged in two stacked rings (blue) of seven subunits each that form two distinct internal polypeptide folding chambers. Homoheptameric lids (10,000-MW subunits), GroES (red), can bind to either end of the barrel and seal the chamber on that side. (b) The GroEL-GroES folding cycle. A partly folded or misfolded polypeptide enters one of the folding chambers (step 1). The second chamber is blocked by a GroES lid. Each ring of seven GroEL subunits binds seven ATPs, hydrolyzes them, and then releases the ADPs in a set order coordinated with GroES binding and release and polypeptide binding, folding, and release. The major conformational changes that take place in the GroEL rings control the binding of the GroES lid that seals the chamber (step 2). The polypeptide remains encased in the chamber capped by the lid, where it can undergo folding until ATP hydrolysis—the slowest, rate-limiting step in the cycle (t½ ~ 10 s) (step 3)—induces binding of ATP and a different GroES to the other ring (transient intermediate shown in brackets). This binding then causes the GroES lid and ADP bound to the peptide-containing ring to be released, opening the chamber and permitting the folded protein to diffuse out of the chamber (step 4). If the polypeptide has folded properly, it can proceed to function in the cell. If it remains partially folded or misfolded, it can rebind to an unoccupied GroEL and the cycle can be repeated. See D. L. Nelson and M. M. Cox, 2013, Lehninger Principles of Biochemistry, 6th ed., Macmillan.
[Part (a) data from Z. Xu, A. L. Horwich, and P. B. Siegler, 1997, Nature388:741–750, PDB ID 1aon.]
Figure 3-18b illustrates the GroEL/GroES cycle of protein folding. A partially folded or misfolded polypeptide of less than 60 kDa in mass is captured by hydrophobic residues near the entrance of the GroEL chamber and enters one of the folding chambers (upper chamber in Figure 3-18b). The second chamber is blocked by a GroES lid. Each of the 14 subunits of GroEL can bind ATP, hydrolyze it, and subsequently release ADP. These reactions are concerted for each set of seven subunits in a single ring and lead to major conformational changes. These changes control both the binding of the GroES lid that seals the chamber and the environment of the chamber in which polypeptide folding takes place. The polypeptide remains encased in the chamber capped by the lid. There it can undergo folding until ATP hydrolysis in that chamber, which is the slowest, rate-limiting step in the cycle (t½ ~10 s), induces binding of ATP and a different GroES to the other ring. This then causes the GroES lid and ADP bound to the peptide-containing ring to be released, opening the chamber and permitting the folded protein to diffuse out of the chamber. If the polypeptide is folded properly, it can proceed to function in the cell. If it remains partially folded or misfolded, it can rebind to an unoccupied GroEL and the cycle can be repeated. There is a reciprocal relationship between the two rings in one GroEL complex. The capping of one chamber by GroES to permit sequestered substrate folding in that chamber is accompanied by the release of substrate polypeptide from the chamber of the second ring (simultaneous binding, folding, and release from the second chamber is not illustrated in Figure 3-18b). There is a striking similarity between the capped-barrel design of GroEL/GroES, in which proteins are sequestered for folding, and the structure of the 26S proteasome that participates in protein degradation (discussed in Section 3.4). In addition, a group of proteins that are part of the AAA+ family of ATPases are composed of hexameric rings with a central pore into which substrates can enter for folding or unfolding or in some cases proteolysis; examples of these will be discussed in Section 3.4 and in Chapter 13.