6.3Examination of Three-Dimensional Structure Enhances Our Understanding of Evolutionary Relationships
Examination of Three-Dimensional Structure Enhances Our Understanding of Evolutionary Relationships
Sequence comparison is a powerful tool for extending our knowledge of protein function and kinship. However, biomolecules generally function as intricate three-
178
Tertiary structure is more conserved than primary structure
Because three-
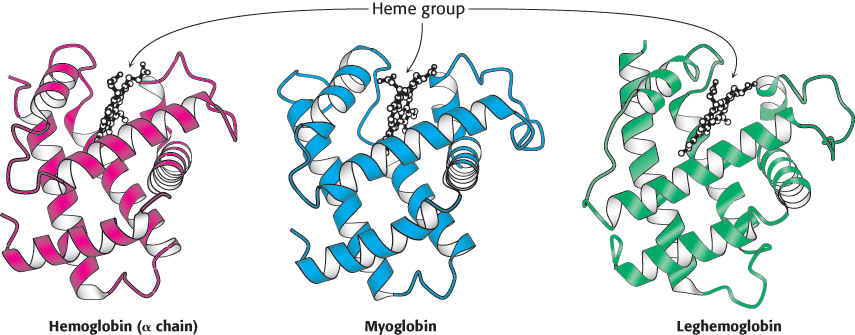
179
Anyone aware of the similar biochemical functions of hemoglobin, myoglobin, and leghemoglobin could expect the structural similarities. In a growing number of other cases, however, a comparison of three-
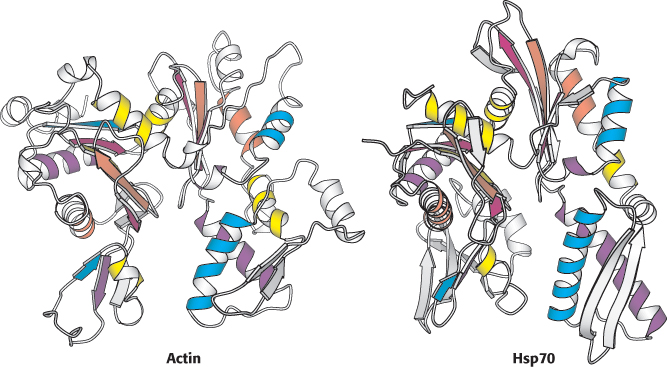
Knowledge of three-dimensional structures can aid in the evaluation of sequence alignments
The sequence-
180
Repeated motifs can be detected by aligning sequences with themselves
More than 10% of all proteins contain sets of two or more domains that are similar to one another. Sequence search methods can often detect internally repeated sequences that have been characterized in other proteins. Often, however, repeated units do not correspond to previously identified domains. In these cases, their presence can be detected by attempting to align a given sequence with itself. The statistical significance of such repeats can be tested by aligning the regions in question as if these regions were sequences from separate proteins. For the TATA-
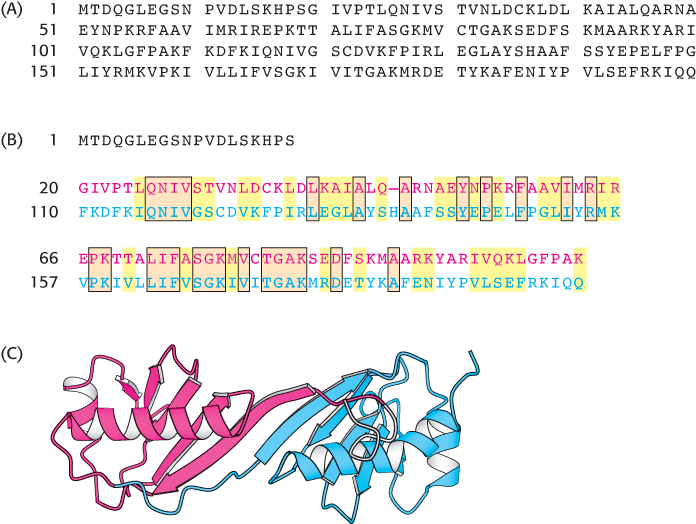
181
Convergent evolution illustrates common solutions to biochemical challenges
Thus far, we have been exploring proteins derived from common ancestors—
An example of convergent evolution is found among the serine proteases. These enzymes, to be considered in more detail in Chapter 9, cleave peptide bonds by hydrolysis. Figure 6.18 shows the structure of the active sites—
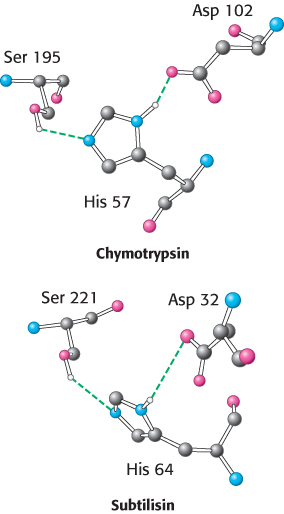
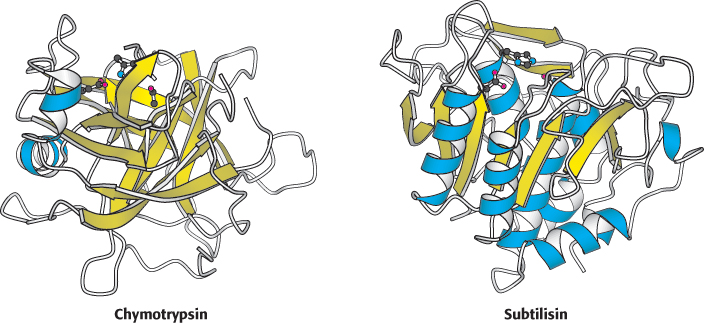
182
Comparison of RNA sequences can be a source of insight into RNA secondary structures
Homologous RNA sequences can be compared in a manner similar to that already described for protein sequences. Such comparisons can be a source of important insights into evolutionary relationships; in addition, they provide clues to the three-
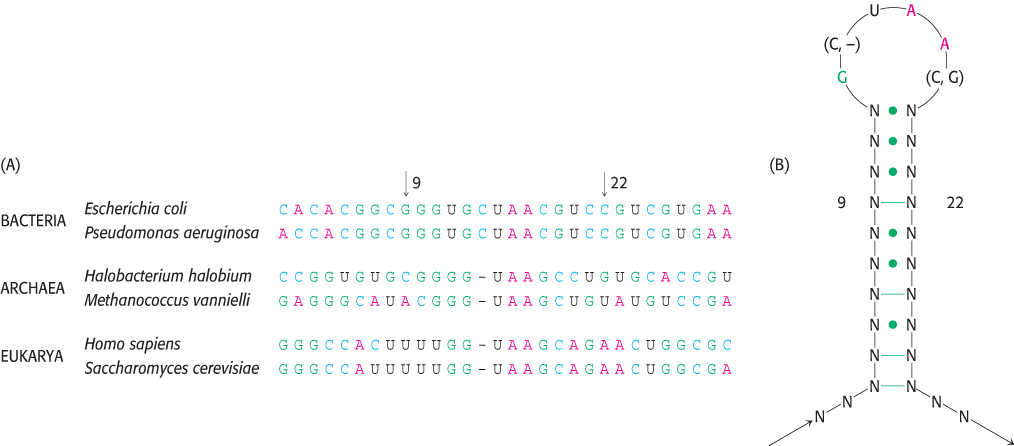
183