21.5
Glycogen Breakdown and Synthesis Are Reciprocally Regulated
An important control mechanism prevents glycogen from being synthesized at the same time as it is being broken down. The same glucagon- and epinephrine-triggered cAMP cascades that initiate glycogen breakdown in the liver and muscle, respectively, also shut off glycogen synthesis. Glucagon and epinephrine control both glycogen breakdown and glycogen synthesis through protein kinase A (Figure 21.19). Recall that protein kinase A adds a phosphoryl group to phosphorylase kinase, activating that enzyme and initiating glycogen breakdown. Glycogen synthase kinase and protein kinase A add phosphoryl groups to glycogen synthase, but this phosphorylation leads to a decrease in enzymatic activity. In this way, glycogen breakdown and synthesis are reciprocally regulated. How is the enzymatic activity reversed so that glycogen breakdown halts and glycogen synthesis begins?
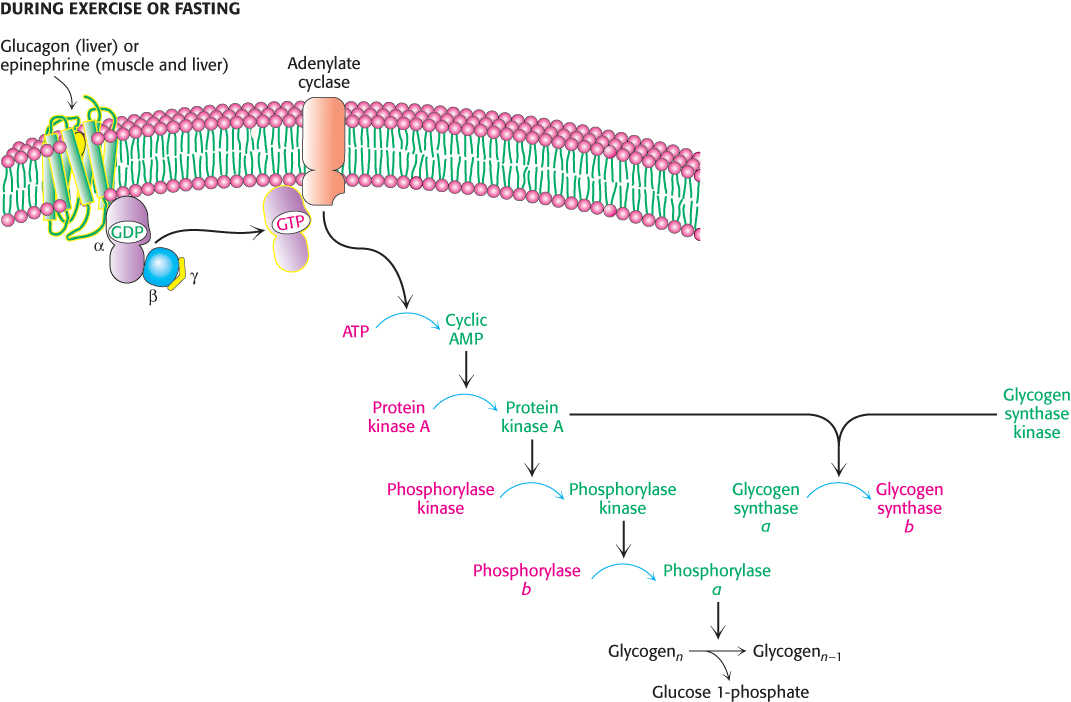
FIGURE 21.19Coordinate control of glycogen metabolism. Glycogen metabolism is regulated, in part, by hormone-triggered cyclic AMP cascades. The sequence of reactions leading to the activation of protein kinase A ultimately activates glycogen degradation. At the same time, protein kinase A along with glycogen synthase kinase inactivates glycogen synthase, shutting down glycogen synthesis.
Protein phosphatase 1 reverses the regulatory effects of kinases on glycogen metabolism
After a bout of exercise, muscle must shift from a glycogen-degrading mode to one of glycogen replenishment. A first step in this metabolic task is to shut down the phosphorylated proteins that stimulate glycogen breakdown. This task is accomplished by protein phosphatases that catalyze the hydrolysis of phosphorylated serine and threonine residues in proteins. Protein phosphatase 1 plays key roles in regulating glycogen metabolism (Figure 21.20). PP1 inactivates phosphorylase a and phosphorylase kinase by dephosphorylating them. PP1 decreases the rate of glycogen breakdown; it reverses the effects of the phosphorylation cascade. Moreover, PP1 also removes phosphoryl groups from glycogen synthase b to convert it into the more active glycogen synthase a form. Here, PP1 also accelerates glycogen synthesis. PP1 is yet another molecular device for coordinating carbohydrate storage. Interestingly, recent research suggests that glycogen phosphorylase is also regulated by acetylation (Section 10.3). Acetylation not only inhbits the enzyme, but also enhances dephosphorylation by promoting the interaction with PP1. Acetylation is stimulated by glucose and insulin, but inhibited by glucagon. It is fascinating that glycogen phosphorylase, one of the first allosteric enzymes identified and one of the most studied enzymes, is still revealing secrets.
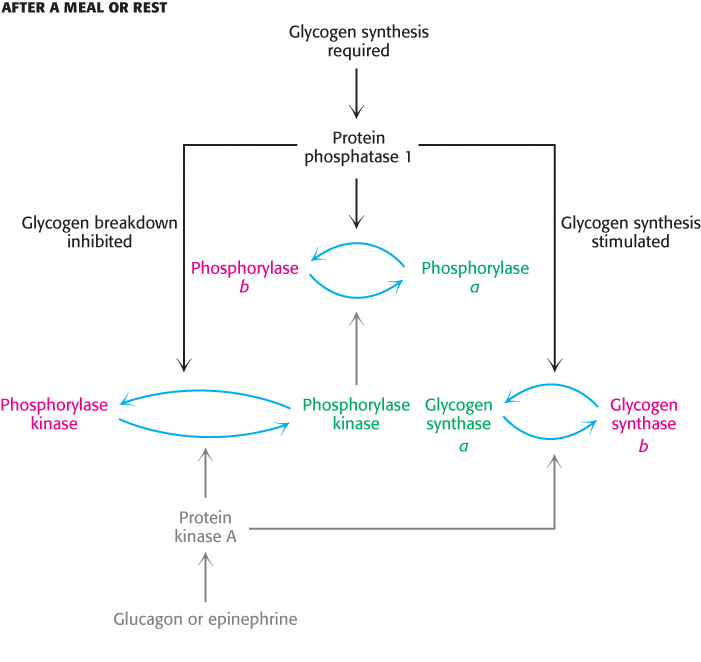
FIGURE 21.20Regulation of glycogen synthesis by protein phosphatase 1. Protein phosphatase 1 stimulates glycogen synthesis while inhibiting glycogen breakdown. Active enzymes are shown in green and inactive enzymes in red.

FIGURE 21.21Regulation of protein phosphatase 1 (PP1) in muscle takes place in two steps. Phosphorylation of GM by protein kinase A dissociates the catalytic subunit from its substrates in the glycogen particle. Phosphorylation of the inhibitor subunit by protein kinase A inactivates the catalytic unit of PP1.
The catalytic subunit of PP1 is a 37-kDa single-domain protein. This subunit is usually bound to one of a family of regulatory subunits with masses of approximately 120 kDa. In skeletal muscle and heart, the most prevalent regulatory subunit is called GM, whereas in the liver, the most prevalent subunit is GL. These regulatory subunits have modular structures with domains that participate in interactions with glycogen, with the catalytic subunit, and with target enzymes. Thus, these regulatory subunits act as scaffolds, bringing together the phosphatase and its substrates on the glycogen particle.
What prevents the phosphatase activity of PP1 from always inhibiting glycogen degradation? When glycogen degradation is called for, epinephrine or glucagon initiate the cAMP cascade that activates protein kinase A (Figure 21.21). Protein kinase A reduces the activity of PP1 by two mechanisms. First, in muscle, GM is phosphorylated in the domain responsible for binding the catalytic subunit. The catalytic subunit is released from glycogen and from its substrates and dephosphoryation is greatly reduced. Second, almost all tissues contain small proteins that, when phosphorylated, bind to the catalytic subunit of PP1 and inhibit it. Thus, when glycogen degradation is switched on by cAMP, the accompanying phosphorylation of these inhibitors keeps phosphorylase in its active a form and glycogen synthase in its inactive b form.
Insulin stimulates glycogen synthesis by inactivating glycogen synthase kinase
After exercise, people often consume carbohydrate-rich foods to restock their glycogen stores. How is glycogen synthesis stimulated? When blood-glucose levels are high, insulin stimulates the synthesis of glycogen by inactivating glycogen synthase kinase, one of the enzymes that maintains glycogen synthase in its phosphorylated, inactive state (Figure 21.22). The first step in the action of insulin is its binding to its receptor, a tyrosine kinase receptor in the plasma membrane (Section 14.2). The binding of insulin stimulates the tyrosine kinase activity of the receptor so that it phosphorylates insulin-receptor substrates (IRSs). These phosphorylated proteins trigger signal-transduction pathways that eventually lead to the activation of protein kinases that phosphorylate and inactivate glycogen synthase kinase. The inactive kinase can no longer maintain glycogen synthase in its phosphorylated, inactive state. Protein phosphatase 1 dephosphorylates glycogen synthase, activating it, and restoring glycogen reserves. Recall that insulin increases the amount of glucose in the cell by increasing the number of glucose transporters in the membrane (Section 16.2). The net effect of insulin is thus the replenishment of glycogen stores.
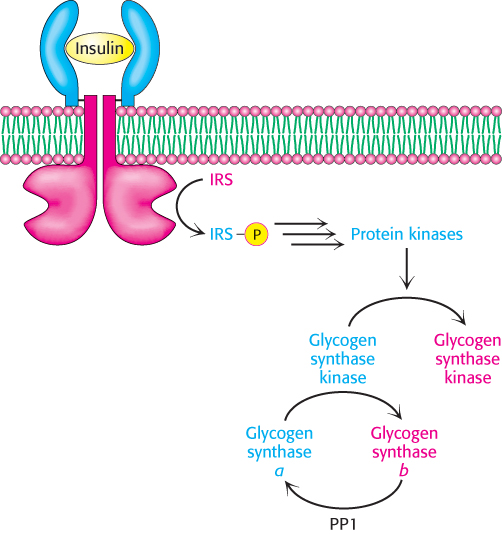
FIGURE 21.22Insulin inactivates glycogen synthase kinase. Insulin triggers a cascade that leads to the phosphorylation and inactivation of glycogen synthase kinase and prevents the phosphorylation of glycogen synthase. Protein phosphatase 1 (PP1) removes the phosphates from glycogen synthase, thereby activating the enzyme and allowing glycogen synthesis. IRS, insulin-receptor substrate.
Glycogen metabolism in the liver regulates the blood-glucose level
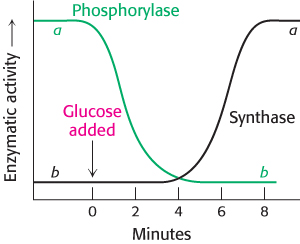
FIGURE 21.23Blood glucose regulates liver-glycogen metabolism. The infusion of glucose into the bloodstream leads to the inactivation of phosphorylase, followed by the activation of glycogen synthase, in the liver.
[Data from W. Stalmans, H. De Wulf, L. Hue, and H.-G. Hers. Eur. J. Biochem. 41:117–134, 1974.]
After a meal rich in carbohydrates, blood-glucose levels rise, and glycogen synthesis is stepped up in the liver. Although insulin is the primary signal for glycogen synthesis, another is the concentration of glucose in the blood, which normally ranges from about 4.4–6.7 mM. The liver senses the concentration of glucose in the blood and takes up or releases glucose accordingly. The amount of liver phosphorylase a decreases rapidly when glucose is infused (Figure 21.23). After a lag period, the amount of glycogen synthase a increases, which results in glycogen synthesis. Phosphorylase a is the glucose sensor in liver cells, facilitating the switch from degradation to synthesis. How does phosphorylase perform its sensor function?
Phosphorylase a and PP1 are localized to the glycogen particle by interactions with the GL subunit of PP1. The binding of glucose to phosphorylase a shifts its allosteric equilibrium from the active R form to the inactive T form. This conformational change renders the phosphoryl group on serine 14 a substrate for protein phosphatase 1. PP1 binds tightly to phosphorylase a only when the phosphorylase is in the R state but is inactive when bound. When glucose induces the transition to the T form, PP1 and the phosphorylase dissociate from each other and the glycogen particle, and PP1 becomes active, converting the phosphorylase a form to the b form. Recall that the R ↔ T transition of muscle phosphorylase a is unaffected by glucose and is thus unaffected by the rise in blood-glucose levels (Section 21.2).
How does glucose binding to glycogen phosphorylase stimulate glycogen synthesis? As mentioned above, the conversion of a into b is accompanied by the release of PP1, which is then free to activate glycogen synthase (Figure 21.24). The removal of the phosphoryl group of inactive glycogen synthase b converts it into the active a form. What accounts for the lag between termination of glycogen degradation and the beginnings of glycogen synthesis (Figure 21.23)? There are about 10 phosphorylase a molecules per molecule of phosphatase. Consequently, the activity of glycogen synthase begins to increase only after most of phosphorylase a is converted into b. The lag between the decrease in glycogen degradation and the increase in glycogen synthesis prevents the two pathways from operating simultaneously. This remarkable glucose-sensing system depends on three key elements: (1) communication between the allosteric site for glucose and the serine phosphate, (2) the use of PP1 to inactivate phosphorylase and activate glycogen synthase, and (3) the binding of the phosphatase to phosphorylase a to prevent the premature activation of glycogen synthase.
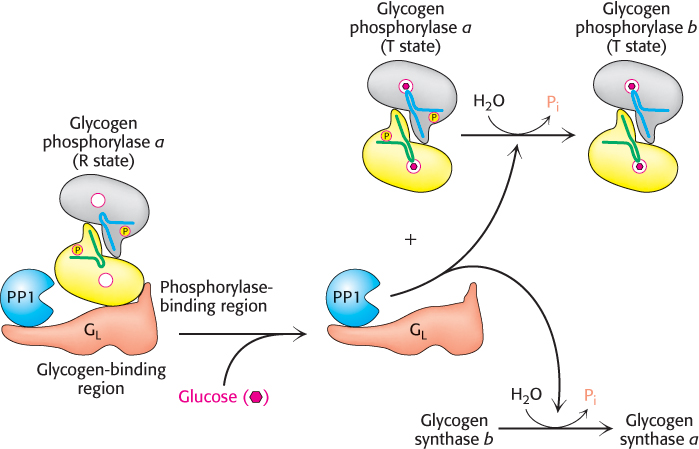
FIGURE 21.24Glucose regulation of liver-glycogen metabolism. Glucose binds to and inhibits glycogen phosphorylase a in the liver, facilitating the formation of the T state of phosphorylase a. The T state of phosphorylase a does not bind protein phosphate 1 (PP1), leading to the dissociation and activation of PP1 from glycogen phosphorylase a. The free PP1 dephosphorylates glycogen phosphorylase a and glycogen synthase b, leading to the inactivation of glycogen breakdown and the activation of glycogen synthesis.
 Efforts are underway to develop drugs that disrupt the interaction of liver phosphorylase with the GL subunit as a treatment for type 2 diabetes (Section 27.2). One experimental drug binds at a unique site on the enzyme that, synergistically with glucose, stabilizes the T state of phosphorylase and thereby enhances the transition to glycogen synthesis, as described earlier. Type 2 diabetes is characterized by excess blood glucose. Hence, disrupting the association of phosphorylase with the GL would render it a substrate for PP1. Glycogen degradation would cease, and glucose release into the blood would be inhibited.
Efforts are underway to develop drugs that disrupt the interaction of liver phosphorylase with the GL subunit as a treatment for type 2 diabetes (Section 27.2). One experimental drug binds at a unique site on the enzyme that, synergistically with glucose, stabilizes the T state of phosphorylase and thereby enhances the transition to glycogen synthesis, as described earlier. Type 2 diabetes is characterized by excess blood glucose. Hence, disrupting the association of phosphorylase with the GL would render it a substrate for PP1. Glycogen degradation would cease, and glucose release into the blood would be inhibited.
A biochemical understanding of glycogen-storage diseases is possible
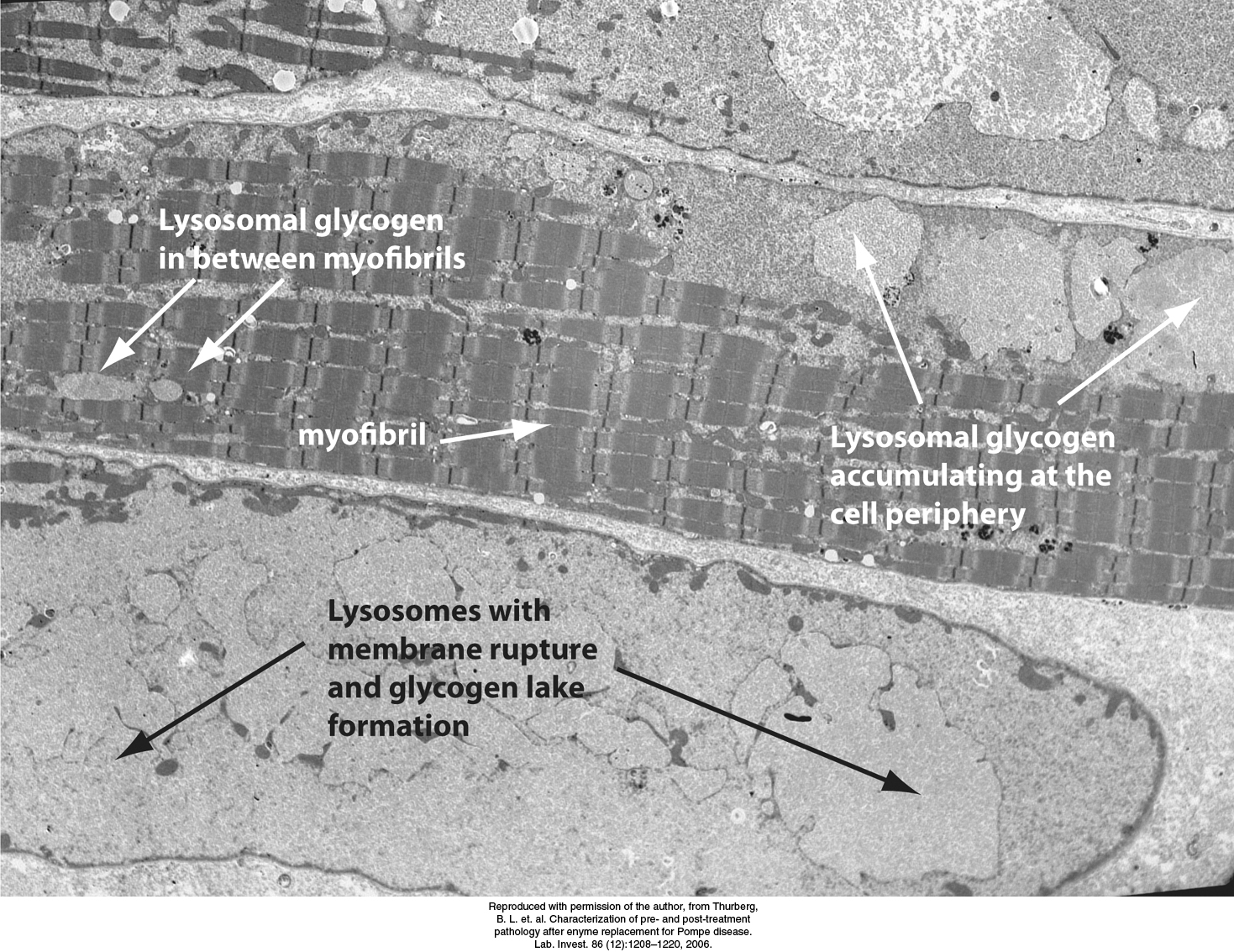
FIGURE 21.25Electron micrograph of muscle cell from a patient with Pompe disease. Glycogen-engorged lysosomes are seen throughout the cell, including the myofibrils. As the disease progresses, lysosomes may rupture, releasing large amounts of glycogen into the cytoplasm. Such accumulations of cytoplasmic glycogen are called glycogen lakes.
[Reproduced with permission of the author, from Thurberg, B. L. et. al. Characterization of pre- and post-treatment pathology after enyme replacement for Pompe disease. Lab. Invest. 86 (12):1208–1220, 2006.]
Edgar von Gierke described the first glycogen-storage disease in 1929. A patient with this disease has a huge abdomen caused by a massive enlargement of the liver. There is a pronounced hypoglycemia between meals. Furthermore, the blood-glucose level does not rise on administration of epinephrine and glucagon. An infant with this glycogen-storage disease may have convulsions because of the low blood-glucose level.
The enzymatic defect in von Gierke disease was elucidated in 1952 by Carl and Gerty Cori. They found that glucose 6-phosphatase is missing from the liver of a patient with this disease. This finding was the first demonstration of an inherited deficiency of a liver enzyme. The glycogen in the liver is normal in structure but is present in abnormally large amounts. The absence of glucose 6-phosphatase in the liver causes hypoglycemia because glucose cannot be formed from glucose 6-phosphate. This phosphorylated sugar does not leave the liver, because it cannot cross the plasma membrane. The presence of excess glucose 6-phosphate triggers an increase in glycolysis in the liver, leading to a high level of lactate and pyruvate in the blood. Patients who have von Gierke disease also have an increased dependence on fat metabolism. This disease can also be produced by a mutation in the gene that encodes the glucose 6-phosphate transporter. Recall that glucose 6-phosphate must be transported into the lumen of the endoplasmic reticulum to be hydrolyzed by phosphatase (Section 16.3). Mutations in the other three essential proteins of this system can likewise lead to von Gierke disease.
Seven other glycogen-storage diseases have been characterized (Table 21.2). In Pompe disease (type II), lysosomes become engorged with glycogen because they lack α-1,4-glucosidase, a hydrolytic enzyme confined to these organelles (Figure 21.25). Carl and Gerty Cori also elucidated the biochemical defect in another glycogen-storage disease (type III), which cannot be distinguished from von Gierke disease (type I) by physical examination alone. In type III disease, the structure of liver and muscle glycogen is abnormal and the amount is markedly increased. Most striking, the outer branches of the glycogen are very short. Patients having this type lack the debranching enzyme (α-1,6-glucosidase), and so only the outermost branches of glycogen can be effectively utilized. Thus, only a small fraction of this abnormal glycogen is functionally active as an accessible store of glucose.
TABLE 21.2 Glycogen-storage diseases
|
|
|
|
Glycogen in the affected organ |
|
|
|
Glucose 6-phosphatase or transport system |
|
Increased amount; normal structure. |
Massive enlargement of the liver; failure to thrive; severe hypoglycemia, ketosis, hyperuricemia, hyperlipemia. |
|
|
α-1,4-Glucosidase (lysosomal) |
|
Massive increase in amount; normal structure. |
Cardiorespiratory failure causes death, usually before age 2. |
|
|
α-1,6-Glucosidase (debranching enzyme) |
|
Increased amount; short outer branches. |
Like type I, but milder course. |
|
|
Branching enzyme (α-1,4 → α-1,6) |
|
Normal amount; very long outer branches. |
Progressive cirrhosis of the liver; liver failure causes death, usually before age 2. |
|
|
|
|
Moderately increased amount; normal structure. |
Limited ability to perform strenuous exercise because of painful muscle cramps; otherwise patient is normal and well developed. |
|
|
|
|
|
Like type I, but milder course. |
|
|
|
|
Increased amount; normal structure. |
|
|
|
|
|
Increased amount; normal structure. |
Mild liver enlargement; mild hypoglycemia. |
Note: Types I through VII are inherited as autosomal recessives. Type VIII is sex linked. |
A defect in glycogen metabolism confined to muscle is found in McArdle disease (type V). Muscle phosphorylase activity is absent, and a patient’s capacity to perform strenuous exercise is limited because of painful muscle cramps. The patient is otherwise normal and well developed. Thus, effective utilization of muscle glycogen is not essential for life.