Like proteins involved in other processes, translation initiation factors and ribosomal proteins can be regulated by post-translational modifications such as phosphorylation. Such mechanisms affect the translation rates of most mRNAs and hence the overall rates of cellular protein synthesis.
TOR Pathway The TOR pathway was discovered through research into the mechanism of action of rapamycin, an antibiotic produced by a strain of Streptomyces bacteria, which is useful for suppressing the immune response in patients who have undergone organ transplants. The target of rapamycin (TOR) was identified by isolating yeast mutants resistant to rapamycin inhibition of cell growth. TOR is a large (~2400-amino-acid) protein kinase that regulates several cellular processes in yeast cells in response to nutritional status. In mammals, mTOR (mammalian TOR) responds to multiple signals from cell-surface signaling proteins to coordinate cell growth with developmental programs as well as with nutritional status.
In mammals, mTOR is assembled into two types of multiprotein complexes, mTOR complexes 1 and 2 (mTORC1 and mTORC2). The protein kinase activity of mTORC1 increases in response to the presence of amino acids in lysosomes. Its protein kinase activity is also increased by levels of ATP sufficient for cell growth, by oxygen, and by signaling from growth-factor receptors (see Chapter 16). mTORC1 is inhibited by various types of cellular stress, including hypoxia and low levels of ATP and nutrients. It is also the mTOR complex inhibited by rapamycin. Active mTORC1 regulates cellular metabolism to promote cell growth and stimulates ribosome synthesis and translation. It also inhibits autophagy, a process in which large portions of the cytoplasm, including whole ribosomes, mitochondria, and other organelles, are surrounded by a double membrane, forming an autophagosome that then fuses with lysosomes, in which the contents are digested to provide essential nutrients in times of stress and when nutrient supply is low. The other complex, mTORC2, is insensitive to rapamycin. When active, it regulates the actin cytoskeleton that controls cell shape and movement (see Chapter 17), and it inhibits apoptosis, a highly organized and regulated pathway to cell death that recycles breakdown products of macromolecules and membranes, making them available for uptake by phagocytic cells (see Chapter 21).
Page 453
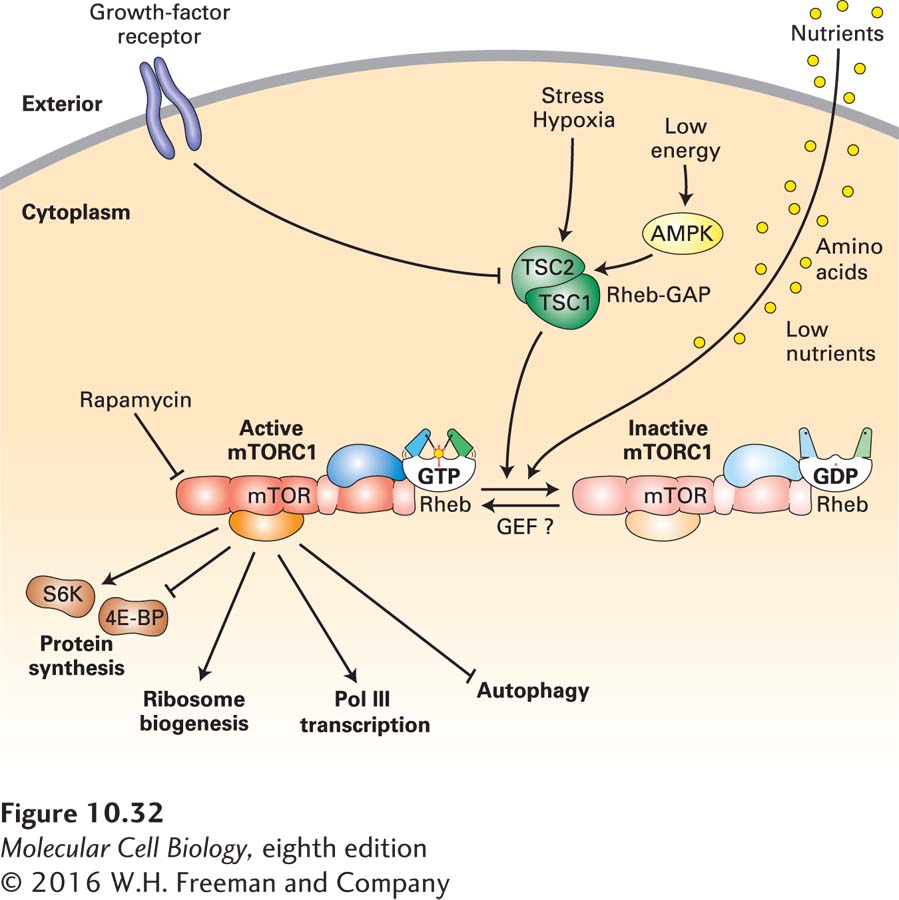
Our current understanding of mTORC1 function is summarized in Figure 10-32. Active mTORC1 increases the overall rate of protein synthesis by phosphorylating two critical types of proteins that regulate translation directly. Recall that the first step in translation of a eukaryotic mRNA is binding of the eIF4 initiation complex to the 5′ cap via its eIF4E cap-binding subunit (see Figure 5-23). The concentration of active eIF4E is regulated by a small family of homologous eIF4E-binding proteins (4E-BPs) that inhibit the interaction of eIF4E with mRNA 5′ caps. 4E-BPs are direct targets of mTORC1. When phosphorylated by mTORC1, 4E-BPs release eIF4E, stimulating translation initiation. mTORC1 also phosphorylates and activates another protein kinase, S6 kinase (S6K), that phosphorylates the small ribosomal subunit protein S6 and additional substrates, leading to a further increase in the rate of protein synthesis.
FIGURE 10-32The mTORC1 pathway. mTORC1 is an active protein kinase when bound by a complex of Rheb and an associated GTP (lower left). In contrast, mTORC1 is inactive when bound by a complex of Rheb associated with GDP (lower right). When active, the TSC1/TSC2 Rheb-GTPase activating protein (Rheb-GAP) causes hydrolysis of Rheb-bound GTP to GDP, thereby inactivating mTORC1. The TSC1/TSC2 Rheb-GAP is activated (arrows) by phosphorylation by AMP kinase (AMPK) when cellular energy is low and by other cellular stress responses. Signal transduction pathways activated by cell-surface growth-factor receptors lead to phosphorylation of inactivating sites on TSC1/TSC2, inhibiting its GAP activity. Consequently, they leave a higher fraction of cellular Rheb in the GTP conformation that activates mTORC1 protein kinase activity. Low nutrient concentrations also regulate Rheb GTPase activity by a mechanism that does not require TSC1/TSC2. Active mTORC1 phosphorylates 4E-BP, causing it to release eIF4E, stimulating translation initiation. It also phosphorylates and activates S6 kinase (S6K), which in turn phosphorylates ribosomal proteins, stimulating translation. Activated mTORC1 also activates transcription factors for RNA polymerases I, II, and III, leading to synthesis and assembly of ribosomes, tRNAs, and translation factors. In the absence of mTORC1 activity, all of these processes are inhibited. In contrast, activated mTORC1 inhibits autophagy, which is stimulated in cells with inactive mTORC1. See S. Wullschleger et al., 2006, Cell124:471.
Translation of a specific subset of mRNAs that have a string of pyrimidines in their 5′ UTR, called TOP mRNAs (for tract of oligopyrimidine), is stimulated particularly strongly by mTORC1. The TOP mRNAs encode ribosomal proteins and translation elongation factors. S6K activated by mTORC1 activates the RNA polymerase I transcription factor TIF-1A, stimulating transcription of the large rRNA precursor (see Figure 9-51). mTORC1 also phosphorylates and inhibits the RNA polymerase III inhibitor MAF1, thereby stimulating synthesis of 5S rRNA and tRNAs. In addition, mTORC1 activates two RNA polymerase II activators that stimulate transcription of genes encoding ribosomal proteins and translation factors. Finally, mTORC1 stimulates processing of the large rRNA precursor (see Section 10.5). As a consequence of the phosphorylation of these several mTORC1 substrates, the synthesis and assembly of ribosomes, as well as the synthesis of translation factors and tRNAs, is greatly increased. Alternatively, when mTORC1 kinase activity is inhibited, these substrates become dephosphorylated, which greatly decreases the rate of protein synthesis and the production of ribosomes, translation factors, and tRNAs, thus halting cell growth.
Page 454
The activity of mTORC1 is regulated by a monomeric G protein in the Ras protein family, called Rheb. Like other small monomeric G proteins, Rheb is in its active conformation when it is bound to GTP (see Figures 15-4 and 15-5). Rheb⋅GTP binds the mTORC1 complex, stimulating mTORC1 kinase activity, probably by inducing a conformational change in its kinase domain. Rheb, in turn, is regulated by a heterodimer composed of subunits TSC1 and TSC2, named for their involvement in the medical syndrome tuberous sclerosis complex, described below. In the active conformation, the TSC1/TSC2 heterodimer functions as a GTPase-activating protein (GAP) for Rheb, causing hydrolysis of the Rheb-bound GTP to GDP. This converts Rheb to its GDP-bound conformation, which does not activate mTORC1 kinase.
The activity of the TSC1/TSC2 Rheb-GAP is regulated by several inputs, allowing the cell to integrate information from different cellular signaling pathways to control the overall rate of protein synthesis. Signaling from cell-surface growth-factor receptors leads to phosphorylation of TSC1/TSC2 at inhibitory sites, causing an increase in Rheb⋅GTP and activation of mTORC1 kinase activity. This type of regulation through cell-surface receptors links the control of cell growth to developmental processes controlled by cell-cell interactions.
When energy from nutrients is not sufficient for cell growth, the resulting fall in the ratio of ATP to AMP concentrations is detected by AMP-activated kinase (AMPK). The activated AMPK phosphorylates TSC1/TSC2 at activating sites, stimulating its Rheb-GAP activity and consequently inhibiting mTORC1 kinase activity and the global rate of translation. Hypoxia and other cellular stresses also activate the TSC1/TSC2 Rheb-GAP.
Activation of mTORC1 depends on the regulated association of mTORC1 with lysosomes. Much of the Rheb in the cell is associated with the outer lysosomal membrane, and other proteins that help Rheb⋅GTP to associate with mTORC1 are restricted to the outer lysosomal membrane. As mentioned previously, regulation of mTORC1 activity is controlled by the lysosomal concentration of amino acids. The mechanism by which this occurs is currently an active area of investigation.
In contrast to mTORC1, mTORC2 is insensitive to nutrients. However, mTORC2 is activated by insulin binding to the insulin receptor, which regulates carbohydrate uptake and metabolism (see Section 16.8). mTORC2 also phosphorylates and activates protein kinase B (also called Akt) (see Figure 16-29), protein kinase C (see page 714), and serum- and glucocorticoid-induced protein kinase 1 (SGK1). These protein kinases, in turn, regulate metabolism, apoptosis, and cell shape through regulation of the actin cytoskeleton (see Chapter 17).
Genes encoding components of the mTORC1 pathway are mutated in many human cancers, resulting in cell growth in the absence of normal growth signals. TSC1 and TSC2 (see Figure 10-32) were initially identified because one or the other is mutated in a rare human genetic syndrome: tuberous sclerosis complex. Patients with this disorder develop benign tumors in multiple tissues. The disease results because inactivation of either TSC1 or TSC2 eliminates the Rheb-GAP activity of the TSC1/TSC2 heterodimer, resulting in an abnormally high and unregulated level of Rheb⋅GTP and thus high, unregulated mTOR activity. Mutations in components of cell-surface receptor signal transduction pathways that lead to inhibition of TSC1/TSC2 Rheb-GAP activity are also common in human tumors and contribute to cell growth and replication in the absence of normal signals for growth and proliferation. High mTORC1 protein kinase activity in tumors correlates with a poor clinical prognosis. Consequently, mTOR inhibitors are currently in clinical trials to test their effectiveness for treating cancers in conjunction with other modes of therapy. Rapamycin and other structurally related mTORC1 inhibitors are potent suppressors of the immune response because they inhibit activation and replication of T lymphocytes in response to foreign antigens (see Chapter 23). Several viruses encode proteins that activate mTORC1 soon after viral infection. The resulting stimulation of translation has an obvious selective advantage for these cellular parasites.
eIF2 Kinases eIF2 kinases also regulate the global rate of cellular protein synthesis. As Figure 5-23 shows, the translation initiation factor eIF2 brings the charged initiator tRNA to the small ribosomal subunit P site. eIF2 is a trimeric G protein and consequently exists in either a GTP-bound or a GDP-bound conformation. Only the GTP-bound form of eIF2 is able to bind the charged initiator tRNA and associate with the small ribosomal subunit. The small ribosomal subunit, with bound initiation factors and charged initiator tRNA, then interacts with the eIF4 complex bound to the 5′ cap of an mRNA via its eIF4E subunit. The small ribosomal subunit then scans down the mRNA in the 3′ direction until it reaches an AUG initiation codon that can base-pair with the initiator tRNA in its P site. When this occurs, the GTP bound by eIF2 is hydrolyzed to GDP and the resulting eIF2⋅GDP complex is released. GTP hydrolysis results in an irreversible “proofreading” step that prepares the small ribosomal subunit to associate with the large subunit only when an initiator tRNA is properly bound in the P site and is properly base-paired with the AUG start codon. Before eIF2 can participate in another round of initiation, its bound GDP must be replaced with a GTP. This process is catalyzed by the translation initiation factor eIF2B, a guanine nucleotide exchange factor (GEF) specific for eIF2.
Page 455
A global mechanism for inhibiting protein synthesis in stressed cells involves phosphorylation of the eIF2α subunit at a specific serine. Phosphorylation at this site does not interfere with eIF2 function in protein synthesis directly. Rather, phosphorylated eIF2 has very high affinity for the eIF2 guanine nucleotide exchange factor, eIF2B, which cannot release the phosphorylated eIF2 and is consequently blocked from catalyzing GTP exchange by additional eIF2 factors. Since there is an excess of eIF2 over eIF2B, phosphorylation of a fraction of eIF2 results in inhibition of all the cellular eIF2B. The remaining eIF2 accumulates in its GDP-bound form, which cannot participate in protein synthesis, thereby inhibiting nearly all protein synthesis in the cell. However, some mRNAs have 5′ regions that allow translation initiation at the low eIF2⋅GTP concentration that results from eIF2 phosphorylation. These mRNAs include those for chaperone proteins that function to refold cellular proteins denatured as the result of cellular stress, additional proteins that help the cell to cope with stress, and transcription factors that activate transcription of the genes encoding these stress-induced proteins.
Humans express four eIF2 kinases that all phosphorylate the same inhibitory eIF2α serine. Each of these kinases is regulated by a different type of cellular stress, and each one inhibits protein synthesis, allowing cells to divert the large fraction of their resources usually devoted to protein synthesis when they are growing for use in responding to the stress.
The GCN2 (general control non-derepressible 2) eIF2 kinase is activated by binding uncharged tRNAs. The concentration of uncharged tRNAs increases when cells are starved for amino acids, activating GCN2 eIF2 kinase and greatly inhibiting protein synthesis.
PEK (pancreatic eIF2 kinase) is activated when proteins translocated into the endoplasmic reticulum (ER) do not fold properly because of abnormalities in the ER lumen environment. Inducers of PEK include abnormal carbohydrate concentrations, which inhibit the glycosylation of many ER proteins. Inactivating mutations in an ER chaperone required for proper folding of many ER proteins (see Chapters 13 and 14) also result in PEK activation.
Heme-regulated inhibitor (HRI) is an eIF2 kinase activated in developing red blood cells when the supply of the heme prosthetic group is too low to accommodate the rate of globin protein synthesis. This negative feedback loop lowers the rate of globin protein synthesis until it matches the rate of heme synthesis. HRI is also activated in other types of cells in response to oxidative stress or heat shock.
Finally, protein kinase RNA-activated (PKR) is activated by double-stranded RNAs longer than about 30 base pairs. Under normal circumstances in mammalian cells, such double-stranded RNAs are produced only during a viral infection. Long regions of double-stranded RNA are generated as replication intermediates of RNA viruses or by hybridization of complementary regions of RNA transcribed from both strands of DNA virus genomes. Inhibition of protein synthesis prevents the production of progeny virions, protecting neighboring cells from infection. Interestingly, adenoviruses have evolved a defense against PKR: they express prodigious amounts of a 160-nucleotide virus-associated (VA) RNA with long double-stranded hairpin regions. VA RNA is transcribed by RNA polymerase III and exported from the nucleus by exportin 5, the exportin for pre-miRNAs (see Figure 10-29). VA RNA binds to PKR with high affinity, inhibiting its protein kinase activity and preventing the inhibition of protein synthesis observed in cells infected with a mutant adenovirus from which the VA gene had been deleted.