An isomerase and a reductase are required for the oxidation of unsaturated fatty acids
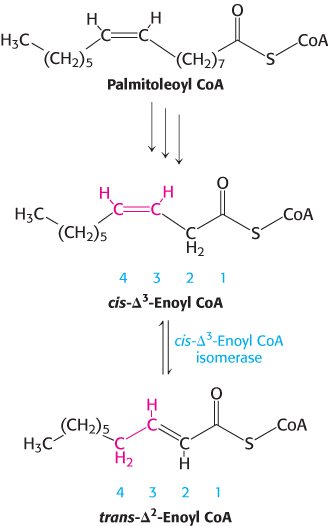
FIGURE 22.11The degradation of a monounsaturated fatty acid. Cis-Δ3-Enoyl CoA isomerase allows continued β-oxidation of fatty acids with a single double bond.
The oxidation of unsaturated fatty acids presents some difficulties, yet many such fatty acids are available in the diet. Most of the reactions are the same as those for saturated fatty acids. In fact, only two additional enzymes—an isomerase and a reductase—are needed to degrade a wide range of unsaturated fatty acids.
Consider the oxidation of palmitoleate (Figure 22.11). This C16 unsaturated fatty acid, which has one double bond between C-9 and C-10, is activated and transported across the inner mitochondrial membrane in the same way as saturated fatty acids are. Palmitoleoyl CoA then undergoes three cycles of degradation, which are carried out by the same enzymes as those in the oxidation of saturated fatty acids. However, the cis-Δ3-enoyl CoA formed in the third round is not a substrate for acyl CoA dehydrogenase. The presence of a double bond between C-3 and C-4 prevents the formation of another double bond between C-2 and C-3. This impasse is resolved by a new reaction that shifts the position and configuration of the cis-Δ3 double bond. cis-Δ3-Enoyl CoA isomerase converts this double bond into a trans-Δ2 double bond. The double bond is now between C-2 and C-3. The subsequent reactions are those of the saturated fatty acid oxidation pathway, in which the trans-Δ2-enoyl CoA is a regular substrate.
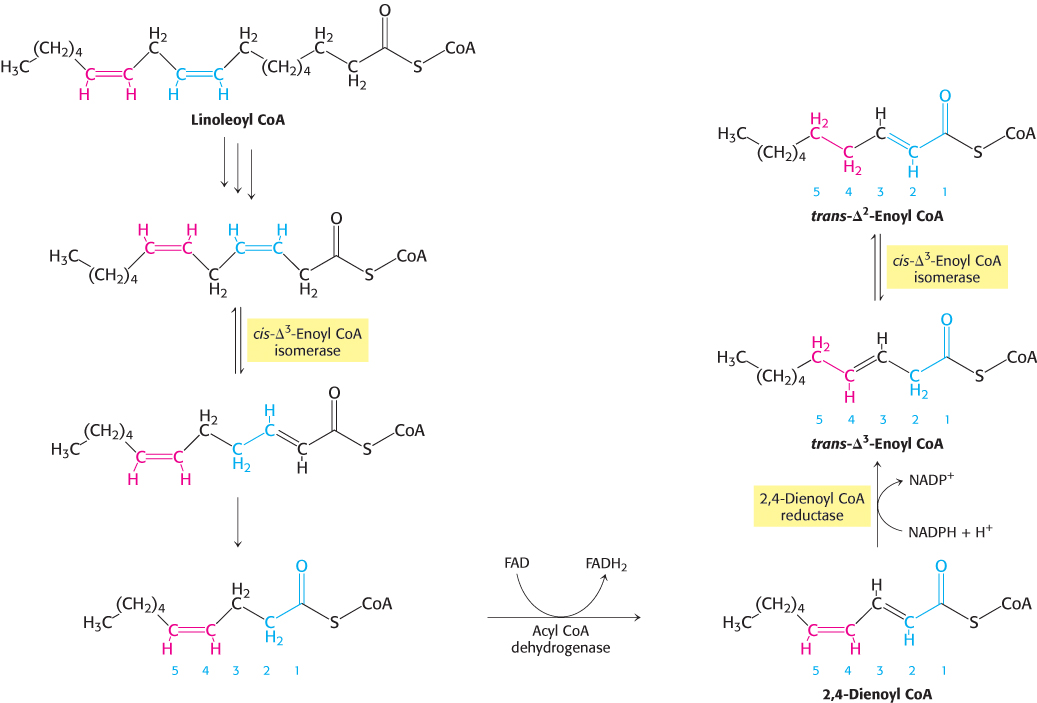
FIGURE 22.12Oxidation of linoleoyl CoA. The complete oxidation of the diunsaturated fatty acid linoleate is facilitated by the activity of enoyl CoA isomerase and 2,4-dienoyl CoA reductase.
Human beings require polyunsaturated fatty acids, which have multiple double bonds, as important precursors for signal molecules, but excess polyunsaturated fatty acids are degraded by β oxidation. However, another problem arises with the oxidation of polyunsaturated fatty acids. Consider linoleate, a C18 polyunsaturated fatty acid with cis-Δ9 and cis-Δ12 double bonds (Figure 22.12). The cis-Δ3 double bond (between carbons 3 and 4) formed after three rounds of β-oxidation is converted into a trans-Δ2 double bond (between carbons 2 and 3) by the aforementioned isomerase. The acyl CoA produced by another round of β-oxidation contains a cis-Δ4 (between carbons 4 and 5) double bond. Dehydrogenation of this species by acyl CoA dehydrogenase yields a 2,4-dienoyl intermediate (double bond between carbons 2 and 3 and carbons 4 and 5), which is not a substrate for the next enzyme in the β-oxidation pathway. This impasse is circumvented by 2,4-dienoyl CoA reductase, an enzyme that uses NADPH to reduce the 2,4-dienoyl intermediate to trans-Δ3-enoyl CoA. cis-Δ3-Enoyl CoA isomerase then converts trans-Δ3-enoyl CoA into the trans-Δ2 form, a customary intermediate in the β-oxidation pathway. These catalytic strategies are elegant and economical. Only two extra enzymes are needed for the oxidation of any polyunsaturated fatty acid. Odd-numbered double bonds are handled by the isomerase, and even-numbered ones by the reductase and the isomerase.
Odd-chain fatty acids yield propionyl CoA in the final thiolysis step
Fatty acids having an odd number of carbon atoms are minor species. They are oxidized in the same way as fatty acids having an even number, except that propionyl CoA and acetyl CoA, rather than two molecules of acetyl CoA, are produced in the final round of degradation. The activated three-carbon unit in propionyl CoA enters the citric acid cycle after it has been converted into succinyl CoA.
The pathway from propionyl CoA to succinyl CoA is especially interesting because it entails a rearrangement that requires vitamin B12 (also known as cobalamin). Propionyl CoA is carboxylated at the expense of the hydrolysis of a molecule of ATP to yield the d isomer of methylmalonyl CoA (Figure 22.13). This carboxylation reaction is catalyzed by propionyl CoA carboxylase, a biotin enzyme that has a catalytic mechanism like that of the homologous enzyme pyruvate carboxylase (Section 16.3). The d isomer of methylmalonyl CoA is racemized to the l isomer, the substrate for a mutase that converts it into succinyl CoA by an intramolecular rearrangement. The —CO—S—CoA group migrates from C-2 to a methyl group in exchange for a hydrogen atom. This very unusual isomerization is catalyzed by methylmalonyl CoA mutase, which contains a derivative of cobalamin as its coenzyme.
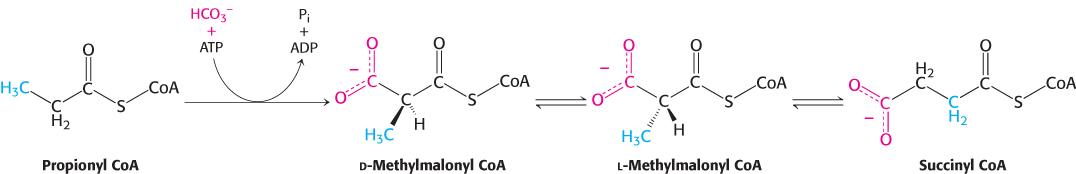
FIGURE 22.13Conversion of propionyl CoA into succinyl CoA. Propionyl CoA, generated from fatty acids with an odd number of carbons as well as some amino acids, is converted into the citric acid cycle intermediate succinyl CoA.
Vitamin B12 contains a corrin ring and a cobalt atom
Cobalamin enzymes, which are present in most organisms, catalyze three types of reactions: (1) intramolecular rearrangements; (2) methylations, as in the synthesis of methionine; and (3) the reduction of ribonucleotides to deoxyribonucleotides (Section 25.3). In mammals, only two reactions are known to require coenzyme B12. The conversion of l-methylmalonyl CoA into succinyl CoA is one, and the formation of methionine by the methylation of homocysteine is the other (Section 24.2). The latter reaction is especially important because methionine is required for the generation of coenzymes that participate in the synthesis of purines and thymine, which are needed for nucleic acid synthesis.
The core of cobalamin consists of a corrin ring with a central cobalt atom (Figure 22.14). The corrin ring, like a porphyrin, has four pyrrole units. Two of them are directly bonded to each other, whereas the others are joined by methine bridges, as in porphyrins. The corrin ring is more reduced than that of porphyrins and the substituents are different. A cobalt atom is bonded to the four pyrrole nitrogens. The fifth substituent linked to the cobalt atom is a derivative of dimethylbenzimidazole that contains ribose 3-phosphate and aminoisopropanol. One of the nitrogen atoms of dimethylbenzimidazole is linked to the cobalt atom. In coenzyme B12, the sixth substituent linked to the cobalt atom is a 5′-deoxyadenosyl unit or a methyl group. This position can also be occupied by a cyano group. Cyanocobalamin is the form of the coenzyme administered to treat B12 deficiency. In all of these compounds, the cobalt is in the +3 oxidation state.
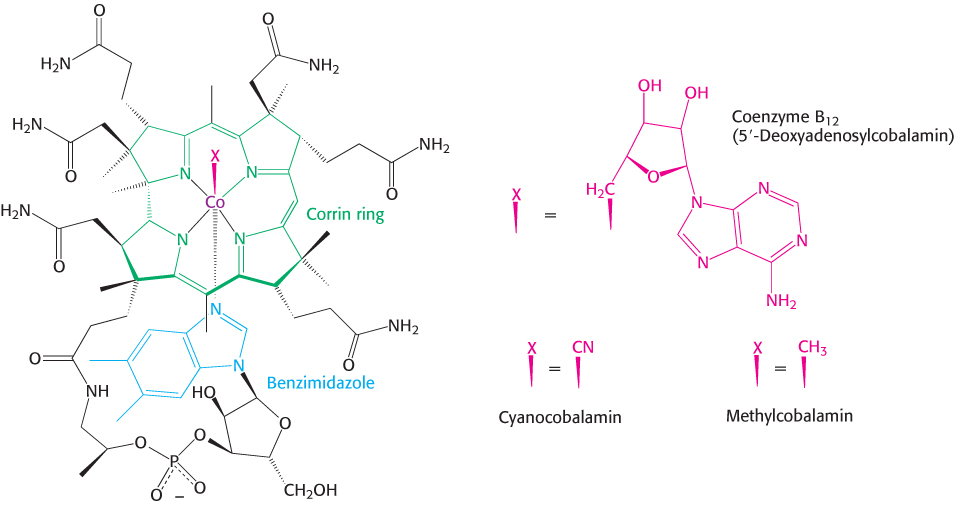
FIGURE 22.14Structure of coenzyme B12. Coenzyme B12 is a class of molecules that vary, depending on the component designated X in the left-hand structure. 5′-Deoxyadenosylcobalamin is the form of the coenzyme in methylmalonyl mutase. Substitution of methyl and cyano groups for X creates methylcobalamin and, cyanocobalamin, respectively.
Mechanism: Methylmalonyl CoA mutase catalyzes a rearrangement to form succinyl CoA
FIGURE 22.15Rearrangement reaction catalyzed by cobalamin enzymes. The R group can be an amino group, a hydroxyl group, or a substituted carbon.
The rearrangement reactions catalyzed by coenzyme B12 are exchanges of two groups attached to adjacent carbon atoms of the substrate (Figure 22.15). A hydrogen atom migrates from one carbon atom to the next, and an R group (such as the —CO—S— CoA group of methylmalonyl CoA) concomitantly moves in the reverse direction. The first step in these intramolecular rearrangements is the cleavage of the carbon–cobalt bond of 5′-deoxyadenosylcobalamin to generate the Co2+ form of the coenzyme and a 5′-deoxyadenosyl radical, —CH2· (Figure 22.16). In this homolytic cleavage reaction, one electron of the Co–C bond stays with Co (reducing it from the +3 to the +2 oxidation state), whereas the other electron stays with the carbon atom, generating a free radical. In contrast, nearly all other cleavage reactions in biological systems are heterolytic: an electron pair is transferred to one of the two atoms that were bonded together.
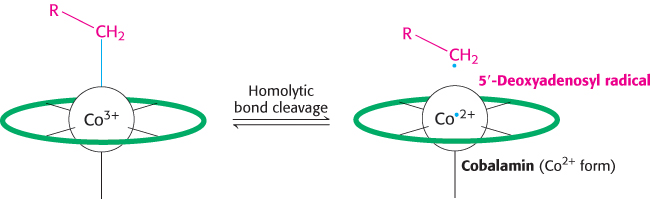
FIGURE 22.16Formation of a 5′-deoxyadenosyl radical. The methylmalonyl CoA mutase reaction begins with the homolytic cleavage of the bond joining Co3+ of coenzyme B12 to a carbon atom of the ribose of the adenosine moiety of the enzyme. The cleavage generates a 5′-deoxyadenosyl radical and leads to the reduction of Co3+ to Co2+. The letter R represents the 5′-deoxyadenosyl component of the coenzyme, and the green oval represents the remainder of the coenzyme.
What is the role of this very unusual —CH2· radical? This highly reactive species abstracts a hydrogen atom from the substrate to form 5′-deoxyadenosine and a substrate radical (Figure 22.17). This substrate radical spontaneously rearranges: the carbonyl CoA group migrates to the position formerly occupied by H on the neighboring carbon atom to produce a different radical. This product radical abstracts a hydrogen atom from the methyl group of 5′-deoxyadenosine to complete the rearrangement and return the deoxyadenosyl unit to the radical form. The role of coenzyme B12 in such intramolecular migrations is to serve as a source of free radicals for the abstraction of hydrogen atoms.
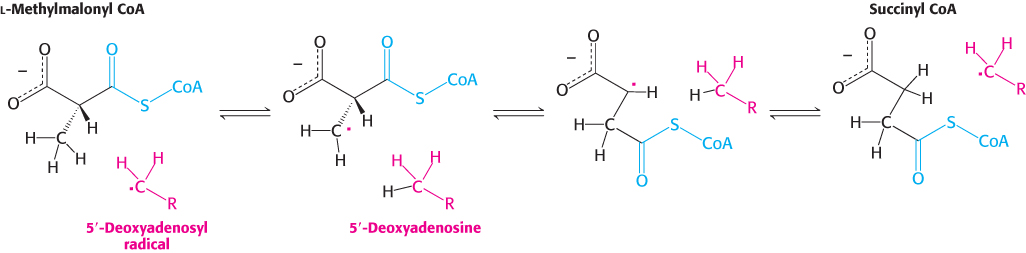
FIGURE 22.17Formation of succinyl CoA by a rearrangement reaction. A free radical abstracts a hydrogen atom in the rearrangement of methylmalonyl CoA to succinyl CoA.
An essential property of coenzyme B12 is the weakness of its cobalt–carbon bond, which is readily cleaved to generate a radical. To facilitate the cleavage of this bond, enzymes such as methylmalonyl CoA mutase displace the benzimidazole group from the cobalamin and bind to the cobalt atom through a histidine residue (Figure 22.18). The steric crowding around the cobalt–carbon bond within the corrin ring system contributes to the bond weakness.
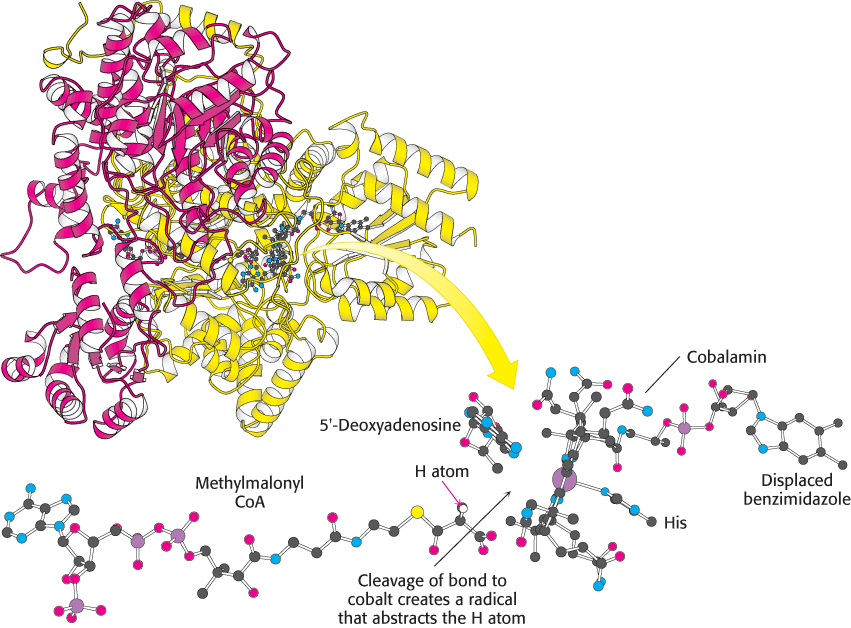
 FIGURE 22.18 Active site of methylmalonyl CoA mutase. Notice that a histidine residue from the enzyme binds to cobalt in place of benzimidazole. This arrangement of substrate and coenzyme in the active site facilitates the cleavage of the cobalt–carbon bond and the subsequent abstraction of a hydrogen atom from the substrate.
FIGURE 22.18 Active site of methylmalonyl CoA mutase. Notice that a histidine residue from the enzyme binds to cobalt in place of benzimidazole. This arrangement of substrate and coenzyme in the active site facilitates the cleavage of the cobalt–carbon bond and the subsequent abstraction of a hydrogen atom from the substrate.
[Drawn from 4REQ.pdb.]
Fatty acids are also oxidized in peroxisomes
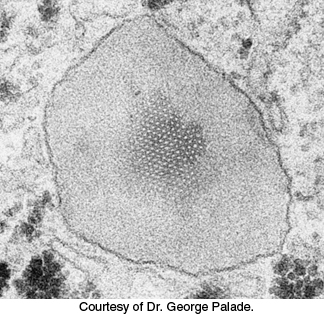
FIGURE 22.19Electron micrograph of a peroxisome in a liver cell. A crystal of urate oxidase is present inside the organelle, which is bounded by a single bilayer membrane. The dark granular structures outside the peroxisome are glycogen particles.
[Courtesy of Dr. George Palade.]
Although most fatty acid oxidation takes place in mitochondria, oxidation of long chain and branched fatty acids takes place in cellular organelles called peroxisomes (Figure 22.19) (problems 17 and 44). These organelles are small membrane-bounded compartments that are present in the cells of most eukaryotes. Fatty acid oxidation in these organelles, which halts at octanoyl CoA, serves to shorten very long chains (C26) to make them better substrates of β oxidation in mitochondria. Peroxisomal oxidation differs from β oxidation in the initial dehydrogenation reaction (Figure 22.20). In peroxisomes, acyl CoA dehydrogenase, a flavoprotein, transfers electrons from the substrate to FADH2 and then to O2 to yield H2O2 instead of capturing high-energy electrons as FADH2 for use in the electron-transport chain, as in mitochondrial β oxidation. Peroxisomes contain high concentrations of the enzyme catalase to degrade H2O2 into water and O2. Subsequent steps are identical with those of their mitochondrial counterparts, although they are carried out by different isoforms of the enzymes.
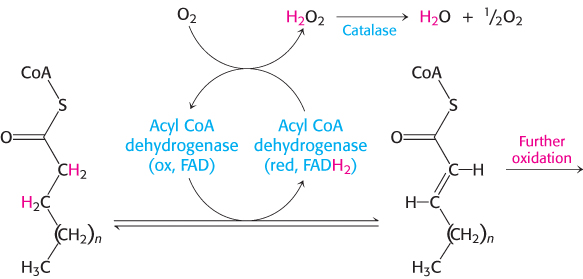
FIGURE 22.20Initiation of peroxisomal fatty acid degradation. The first dehydrogenation in the degradation of fatty acids in peroxisomes requires a flavoprotein dehydrogenase that transfers electrons from its FADH2 moiety to O2 to yield H2O2.
 Peroxisomes do not function in patients with Zellweger syndrome. Liver, kidney, and muscle abnormalities usually lead to death by age 6. The syndrome is caused by a defect in the import of enzymes into the peroxisomes. Here we see a pathological condition resulting from an inappropriate cellular distribution of enzymes.
Peroxisomes do not function in patients with Zellweger syndrome. Liver, kidney, and muscle abnormalities usually lead to death by age 6. The syndrome is caused by a defect in the import of enzymes into the peroxisomes. Here we see a pathological condition resulting from an inappropriate cellular distribution of enzymes.
Ketone bodies are formed from acetyl CoA when fat breakdown predominates
The acetyl CoA formed in fatty acid oxidation enters the citric acid cycle only if fat and carbohydrate degradation are appropriately balanced. Acetyl CoA must combine with oxaloacetate to gain entry to the citric acid cycle. The availability of oxaloacetate, however, depends on an adequate supply of carbohydrate. Recall that oxaloacetate is normally formed from pyruvate, the product of glucose degradation in glycolysis, by pyruvate carboxylase (Section 16.3). If carbohydrate is unavailable or improperly utilized, the concentration of oxaloacetate is lowered and acetyl CoA cannot enter the citric acid cycle. This dependency is the molecular basis of the adage that fats burn in the flame of carbohydrates.
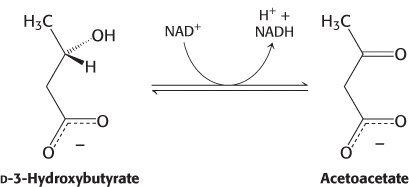
In fasting or diabetes, oxaloacetate is consumed to form glucose by the gluconeogenic pathway (Section 16.3) and hence is unavailable for condensation with acetyl CoA. Under these conditions, acetyl CoA is diverted to the formation of acetoacetate and d-3-hydroxybutyrate. Acetoacetate, d-3-hydroxybutyrate, and acetone are often referred to as ketone bodies. Abnormally high levels of ketone bodies are present in the blood of untreated diabetics.
Acetoacetate is formed from acetyl CoA in three steps (Figure 22.21). Two molecules of acetyl CoA condense to form acetoacetyl CoA. This reaction, which is catalyzed by thiolase, is the reverse of the thiolysis step in the oxidation of fatty acids. Acetoacetyl CoA then reacts with acetyl CoA and water to give 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) and CoA. This condensation resembles the one catalyzed by citrate synthase (Section 17.2). This reaction, which has a favorable equilibrium owing to the hydrolysis of a thioester linkage, compensates for the unfavorable equilibrium in the formation of acetoacetyl CoA. 3-Hydroxy-3-methylglutaryl CoA is then cleaved to acetyl CoA and acetoacetate. The sum of these reactions is
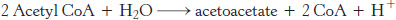
FIGURE 22.21Formation of ketone bodies. The ketone bodies—acetoacetate, d-3-hydroxybutyrate, and acetone—are formed from acetyl CoA primarily in the liver. Enzymes catalyzing these reactions are (1) 3-ketothiolase, (2) hydroxymethylglutaryl CoA synthase, (3) hydroxymethylglutaryl CoA cleavage enzyme, and (4) d-3-hydroxybutyrate dehydrogenase. Acetoacetate spontaneously decarboxylates to form acetone.
d-3-Hydroxybutyrate is formed by the reduction of acetoacetate in the mitochondrial matrix by d-3-hydroxybutyrate dehydrogenase. The ratio of hydroxybutyrate to acetoacetate depends on the NADH/NAD+ ratio inside mitochondria.
Because it is a β-ketoacid, acetoacetate also undergoes a slow, spontaneous decarboxylation to acetone. The odor of acetone may be detected in the breath of a person who has a high level of acetoacetate in the blood. Under starvation conditions, the acetone may be captured to synthesize glucose.
Ketone bodies are a major fuel in some tissues
The major site of the production of acetoacetate and 3-hydroxybutyrate is the liver. These molecules diffuse from the liver mitochondria into the blood and are transported to other tissues such as heart and kidney (Figure 22.22). Acetoacetate and 3-hydroxybutyrate are normal fuels of respiration and are quantitatively important as sources of energy. Indeed, heart muscle and the renal cortex use acetoacetate in preference to glucose. In contrast, glucose is the major fuel for the brain and red blood cells in well-nourished people on a balanced diet. However, the brain adapts to the utilization of acetoacetate during starvation and diabetes. In prolonged starvation, 75% of the fuel needs of the brain are met by ketone bodies.
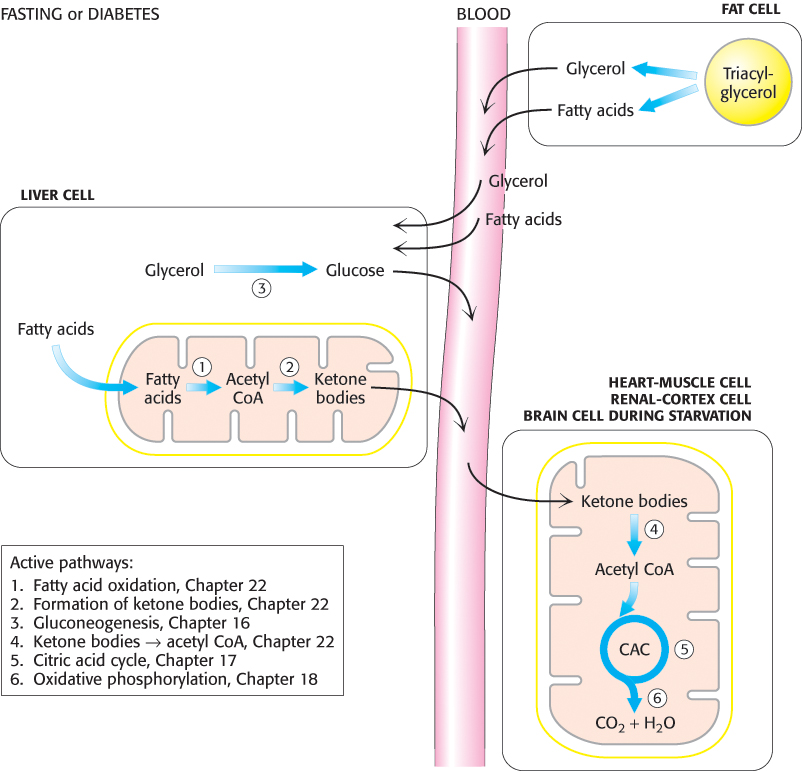
FIGURE 22.22PATHWAY INTEGRATION: Liver supplies ketone bodies to the peripheral tissues. During fasting or in untreated diabetics, the liver converts fatty acids into ketone bodies, which are a fuel source for a number of tissues. Ketone-body production is especially important during starvation, when ketone bodies are the predominant fuel.
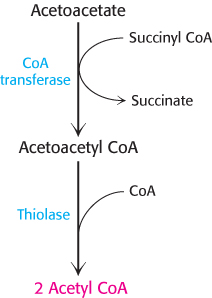
FIGURE 22.23Utilization of acetoacetate as a fuel. Acetoacetate can be converted into two molecules of acetyl CoA, which then enter the citric acid cycle.
Acetoacetate is converted into acetyl CoA in two steps. First, acetoacetate is activated by the transfer of CoA from succinyl CoA in a reaction catalyzed by a specific CoA transferase. Second, acetoacetyl CoA is cleaved by thiolase to yield two molecules of acetyl CoA, which can then enter the citric acid cycle (Figure 22.23). The liver has acetoacetate available to supply to other organs because it lacks this particular CoA transferase. 3-Hydroxybutyrate requires an additional step to yield acetyl CoA. It is first oxidized to produce acetoacetate, which is processed as heretofore described, and NADH for use in oxidative phosphorylation.
Ketone bodies can be regarded as a water-soluble, transportable form of acetyl units. Fatty acids are released by adipose tissue and converted into acetyl units by the liver, which then exports them as acetoacetate. As might be expected, acetoacetate also has a regulatory role. High levels of acetoacetate in the blood signify an abundance of acetyl units and lead to a decrease in the rate of lipolysis in adipose tissue.
High blood levels of ketone bodies, the result of certain pathological conditions, can be life threatening. The most common of these conditions is diabetic ketosis in patients with insulin-dependent diabetes mellitus. These patients are unable to produce insulin. As stated earlier, this hormone, normally released after meals, signals tissues to take up glucose. In addition, it curtails fatty acid mobilization by adipose tissue. The absence of insulin has two major biochemical consequences (Figure 22.24). First, the liver cannot absorb glucose and consequently cannot provide oxaloacetate to process fatty acid-derived acetyl CoA. Second, adipose cells continue to release fatty acids into the blood stream; these fatty acids are then taken up by the liver and converted into ketone bodies. The liver thus produces large amounts of ketone bodies, which are moderately strong acids. The result is severe acidosis. The decrease in pH impairs tissue function, most importantly in the central nervous system.
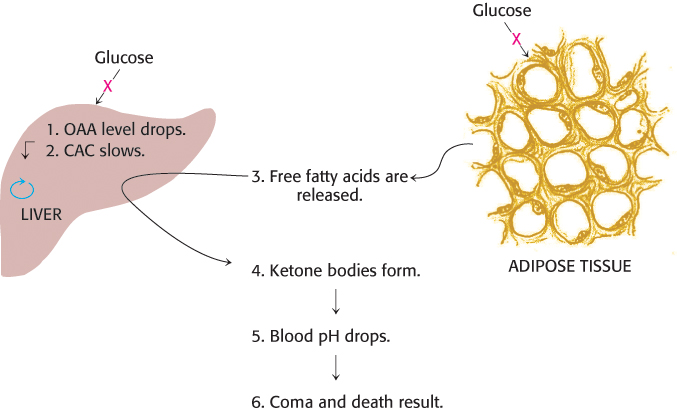
FIGURE 22.24Diabetic ketosis results when insulin is absent. In the absence of insulin, fats are released from adipose tissue, and glucose cannot be absorbed by the liver or adipose tissue. The liver degrades the fatty acids by β oxidation but cannot process the acetyl CoA, because of a lack of glucose-derived oxaloacetate (OAA). Excess ketone bodies are formed and released into the blood.
Interestingly, diets that promote ketone-body formation, called ketogenic diets, are frequently used as a therapeutic option for children with drug-resistant epilepsy. Ketogenic diets are rich in fats and low in carbohydrates, with adequate amounts of protein. In essence, the body is forced into starvation mode, where fats and ketone bodies become the main fuel source (Section 27.5). How such diets reduce the seizures suffered by the children is currently unknown.
Animals cannot convert fatty acids into glucose
A typical human being has far greater fat stores than glycogen stores. However, glycogen is necessary to fuel very active muscle, as well as the brain, which normally uses only glucose as a fuel. When glycogen stores are low, why can’t the body make use of fat stores and convert fatty acids into glucose? Because animals are unable to effect the net synthesis of glucose from fatty acids. Specifically, acetyl CoA cannot be converted into pyruvate or oxaloacetate in animals. Recall that the reaction that generates acetyl CoA from pyruvate is irreversible (Section 17.1). The two carbon atoms of the acetyl group of acetyl CoA enter the citric acid cycle, but two carbon atoms leave the cycle in the decarboxylations catalyzed by isocitrate dehydrogenase and α-ketoglutarate dehydrogenase. Consequently, oxaloacetate is regenerated, but it is not formed de novo when the acetyl unit of acetyl CoA is oxidized by the citric acid cycle. In essence, two carbon atoms enter the cycle as an acetyl group, but two carbons leave the cycle as CO2 before oxaloacetate is generated. As a result, no net synthesis of oxaloacetate is possible. In contrast, plants have two additional enzymes enabling them to convert the carbon atoms of acetyl CoA into oxaloacetate (Section 17.5).
Some fatty acids may contribute to the development of pathological conditions
As we will see shortly (Section 22.5), certain polyunsaturated fatty acids are essential for life, serving as precursors to various signal molecules. Vegetable oils, used commonly in food preparation, are rich in polyunsaturated fatty acids. However, polyunsaturated fatty acids are unstable and are readily oxidized. This tendency to become rancid reduces their shelf life and renders them undesirable for cooking. To circumvent this problem, polyunsaturated fatty acids are hydrogenated, converting them to saturated and trans unsaturated fatty acids (popularly known as “trans fat”), a variety of fat that is rare in nature. Epidemiological evidence suggests that consumption of large amounts of saturated fatty acids and trans fat promotes obesity, type 2 diabetes, and atherosclerosis. The mechanism by which these fats exert these effects is under active investigation. Some evidence suggests that they promote an inflammatory response and may mute the action of insulin and other hormones (Section 27.3).