There Are Four Broad Structural Categories of Proteins
Proteins usually fall into one of four broad structural categories based on their tertiary structure: globular proteins, fibrous proteins, integral membrane proteins, and intrinsically disordered proteins. These four broad categories of proteins are not mutually exclusive—some proteins are made up of combinations of segments that fall into two or more of these categories. Globular proteins are generally water-soluble, compactly folded structures, often but not exclusively spheroidal, that comprise a mixture of secondary structures [see the structures of ras (Figure 3-9 below) and myoglobin (Figure 3-14 below)]. Fibrous proteins are large, elongated, often stiff molecules. Some fibrous proteins are composed of a long polypeptide chain comprising many tandem copies of a short amino acid sequence that forms a single repeating secondary structure (see the structure of collagen, the most abundant protein in mammals, in Figure 20-25). Other fibrous proteins are composed of repeating globular protein subunits, such as the helical array of G-actin protein monomers that forms F-actin microfilaments (see Chapter 17). Fibrous proteins, which often aggregate into large multiprotein fibers that do not readily dissolve in water, usually play a structural role or participate in cellular movements. Integral membrane proteins are embedded within the phospholipid bilayer of the membranes that enclose cells and organelles and are discussed in detail in Chapter 7.
Page 73
Intrinsically disordered proteins are fundamentally distinct from the well-ordered proteins in the other three categories. Many proteins we consider in this book adopt only one or a few very closely related conformations when they are in their normal functional state, called the native state. Intrinsically disordered proteins, however, do not have well-ordered structures in their native, functional states; instead, their polypeptide chains are very flexible—indeed, disordered—with no fixed conformation. Sometimes only a segment of a polypeptide chain, rather than the entire chain, will be intrinsically disordered. The exceptional conformational flexibilities of intrinsically disordered proteins or protein segments appear to be key to their functional activities, such as the ability to interact with multiple partner proteins or to fold into a well-defined conformation only after binding to such partners (Figure 3-8a).
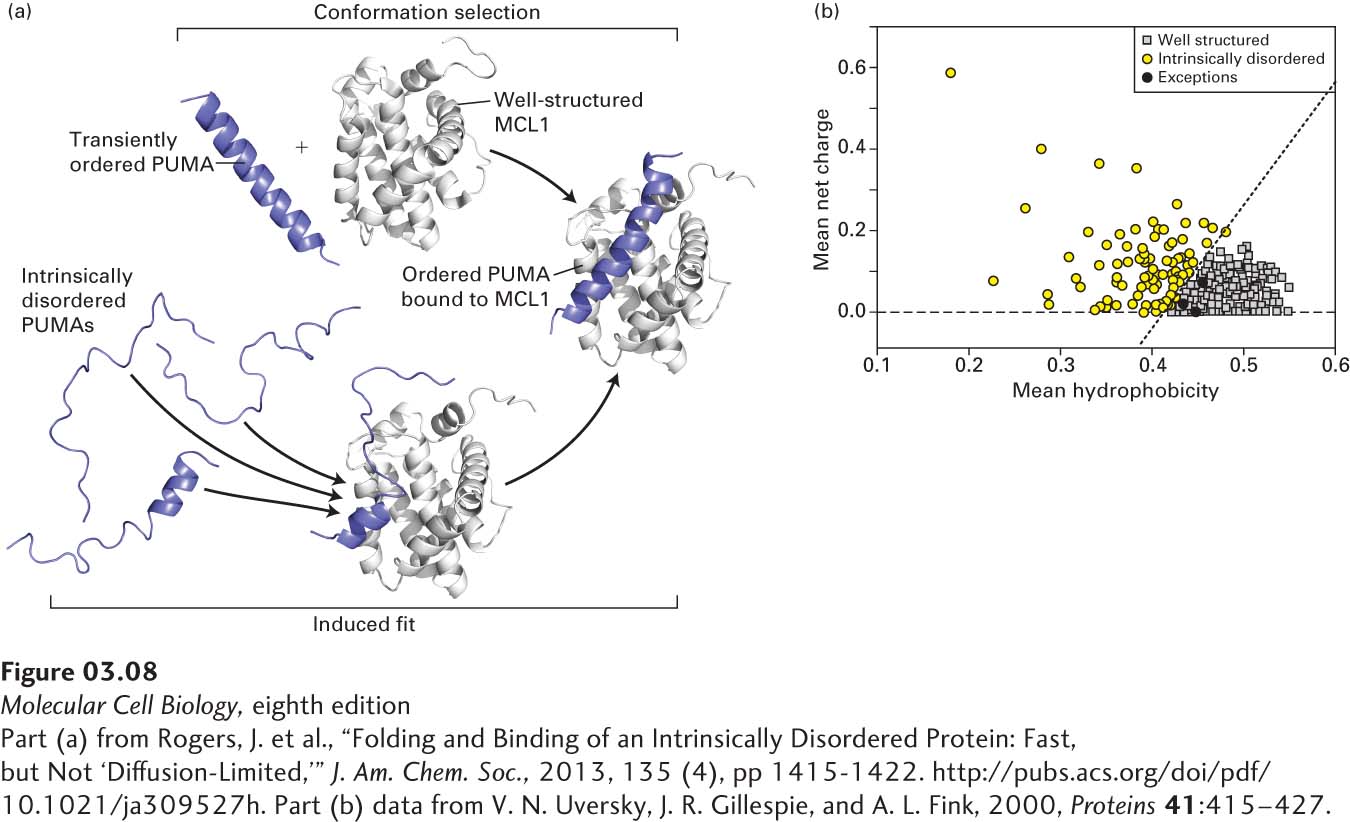
[Part (a) from Rogers, J. et al., “Folding and Binding of an Intrinsically Disordered Protein: Fast, but Not ‘Diffusion-Limited,’” J. Am. Chem. Soc., 2013, 135 (4), pp1415-1422. http://pubs.acs.org/doi/pdf/10.1021/ja309527h. Part (b) data from V. N. Uversky, J. R. Gillespie, and A. L. Fink, 2000, Proteins41:415–427.]
EXPERIMENTAL FIGURE 3-8Intrinsically disordered proteins: mechanisms of binding to well-ordered proteins and identification based on hydrophobicity and net charge. (a) The binding of an intrinsically disordered protein (PUMA, blue) to a well-ordered protein (MCL1, gray) results in the formation of a well-defined structure in the previously disordered protein. PUMA and MCL1 are intracellular proteins that can influence the regulated process of cell death called apoptosis (see Chapter 21). Two mechanisms have been proposed for generating a bound complex in which both proteins are structured: conformational selection (top pathway) and induced fit (bottom pathway). In conformational selection, the disordered protein (PUMA) occasionally and transiently adopts in solution the structure it would have in the bound state. The well-ordered binding partner (MLC1) can then bind to (select) PUMA in that transient, ordered conformation, forming a relatively stable bound complex. In induced fit, the disordered protein begins to bind to the well-ordered partner while still disordered and then, while bound, is induced to form the ordered conformation present in the relatively stable, heterodimeric complex. Recent experiments suggest that the induced fit mechanism best describes the binding of PUMA and MCL1. (b) The sequences of 275 well-ordered, monomeric globular proteins (gray squares) and 91 intrinsically disordered proteins (black and yellow circles) were used to calculate the mean hydrophobicity per residue in each protein using a scale of 0 (least hydrophobic) to 1 (most hydrophobic, x axis), and the mean net charge per residue at pH 7.0 (y axis). With only three exceptions (black circles), the proteins define two distinct distributions: low hydrophobicity, high net charge (intrinsically disordered, yellow circles) and high hydrophobicity, low net charge (well-ordered, gray squares). The three disordered proteins (black circles) that overlap with the well-ordered population each contain substantial segments predicted to be disordered (low hydrophobicity, high net charge) that apparently overwhelm the rest of the proteins’ sequences that might otherwise result in a well-ordered conformation.
[Part (a) from Rogers, J. et al., “Folding and Binding of an Intrinsically Disordered Protein: Fast, but Not ‘Diffusion-Limited,’” J. Am. Chem. Soc., 2013, 135 (4), pp1415-1422. http://pubs.acs.org/doi/pdf/10.1021/ja309527h. Part (b) data from V. N. Uversky, J. R. Gillespie, and A. L. Fink, 2000, Proteins41:415–427.]
Intrinsically disordered proteins typically, but not exclusively, serve as signaling molecules, regulators of the activities of other molecules, or as scaffolds for multiple proteins, small molecules, and ions (e.g., binding ions via multiple charged residues). Regions of intrinsic disorder can provide flexible links, or tethers, between well-ordered regions of a protein; serve as sites of some types of post-translational protein modification [e.g., covalent addition of phosphate groups (phosphorylation) or sugars (glycosylation)]; serve as targets of protease digestion that regulates protein activity; inhibit the activity of the protein in which they are embedded (autoinhibition sites); or serve as signals for intracellular sorting of proteins (see Chapter 13). The activities of many proteins containing intrinsically disordered segments are described in subsequent chapters. For example, phosphorylation of the disordered C-terminal domain (CTD) of RNA polymerase II (see Figure 8-12), which is composed of multiple repeats of a seven-amino-acid sequence containing proline, threonine, and serine, regulates key steps in the synthesis of mRNA (see Chapters 9 and 10). The N-termini of histone proteins that control DNA organization in chromatin (see Chapter 8) are sites of important post-translational modifications, and the disordered, proline-rich FH1 region in the protein formin controls the assembly of actin filaments (see Chapter 17).
Intrinsically disordered proteins can be identified experimentally using various biochemical techniques, such as tests of sensitivity to protease digestion (disordered regions usually exhibit greater protease sensitivity), and a wide variety of biophysical techniques, including spectroscopy. The intrinsic disorder of these proteins apparently arises as a consequence of their having a sequence that, relative to well-ordered proteins, is richer in polar amino acids, proline, and net charge, and poorer in hydrophobic residues (Figure 3-8b). Algorithms primarily based on calculations of amino acid composition—particularly net charge and hydrophobicity—are used to predict which proteins or segments of proteins are intrinsically disordered. By some estimates, about 30 percent or more of eukaryotic proteins are predicted to have at least one segment of 50 or more consecutive residues that is disordered.